 "
"
Team:Heidelberg/Project/Tag-Optimization
From 2013.igem.org



Project

Notebook



Human Practice




Tag-Optimization. Engineering indC by Domain Exchanges.

Highlights
Abstract
Non-ribosomal peptide synthetases (NRPS) offer a unique opportunity to spin around their inherent logical assembly and observe if their functionality is preserved or even improved.
Following this idea, we investigate the interchangeability of NRPS domains and the possibility to tune their efficiency at the example of indC from Photorhabdus luminescens, the NRPS module used for the Indigoidine-Tag. The native NRPS domains have been replaced with domains from other bacterial organisms and fully synthetic domains. Moreover, we compare the activity of different PPTases, which are required for the activation of NRPS modules. To determine the NRPS efficiency we established a quantitative indigoidine assay based on OD measurement of the blue-colored pigment. Interestingly, our data points out that some of our engineered indC variants exhibit increased efficiency in producing indigoidine compared to the native enzyme. Furthermore, we introduce HiCT - High throughput protocols for circular polymerase extension Cloning and Transformation - a new standard for the assembly of combinatorial gene libraries (RFC 99).








Introduction
In Non-Ribosomal Peptide Synthesis, peptides are produced by multienzyme complexes, the NRPS, which form an assembly line, in which every NRPS module is responsible for the incorporation of one single amino acid into the growing peptide chain (for more detailled information on the NRPS system, please visit our Background Page to see our NRPS introduction video)[1]. As previously shown, the natural NRPS assembly lines can be rearranged to create novel assembly lines producing custom peptides (please find our experiments on the Peptide Synthesis Page)[2]. Since the detection of those custom peptides remained challenging when produced in vivo, we developped a method which enables the labelling of non-ribosomal peptides (NRPs) with the blue pigment indigoidine (please find our experiments on the Indigoidine-Tag Page). The Indigoidine-Tag enables for applying high-throughput methods in the creation of NRPS libraries as well as in detection, purification and validation of synthetic NRPs (please find our RFC Page for standardized high-throughput protocols). Custom short peptides might have a great potential for production in industrial scale. Therefore, we try to optimize the efficiency of the indigoidine synthetase indC, which is used for the Indigoidine-Tag.
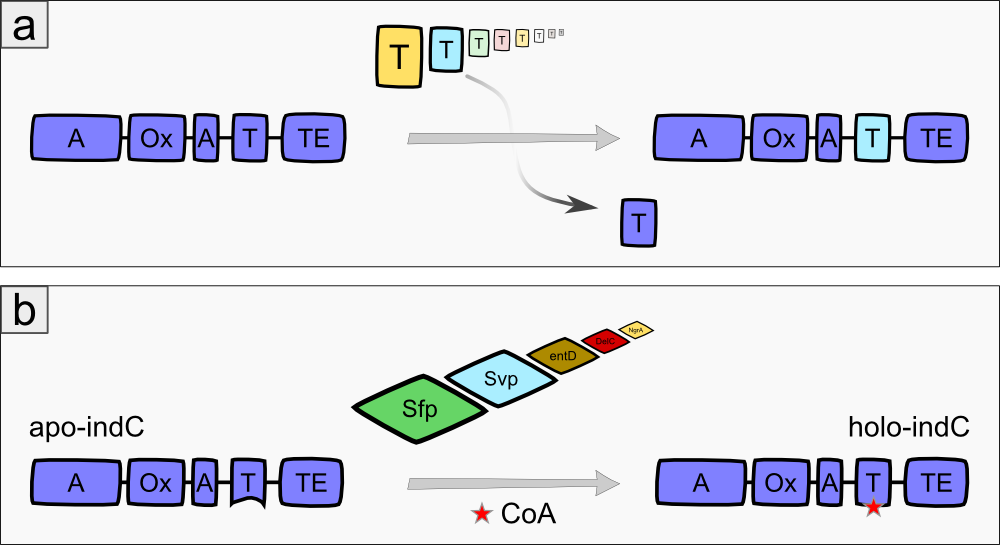
The indigoidine synthetase indC from Photorhabdus luminescens laumondii TT01 (DSM15139) consists of an adenylation domain with an internal oxidation domain, a thiolation domain and a thioesterase domain [3]. The A-domain adenylates L-glutamine, which is then attached to the T-domain via a thioester bond. The TE-domain catalyzes the cyclization of the glutamine and cleaves it from the T-domain. Each two cyclic glutamines are oxidized by the Ox-domain, resulting in the blue pigment indigoidine (Fig. 1a). The indigoidine synthetase must be activated by an enzyme called 4'Phosphopantheteinyl-transferase (PPTase), which transfers the 4'-PPT residue from Coenzyme A to a conserved serine residue in the T-domain of the indigoidine synthetase, thus transforming it from its inactive apo- to the active holo-form (Fig. 1b)[3].

We expressed the indigoidine synthetase indC together with the PPTase in different substrains of E. coli to observe their growth behaviour and to get an impression, which substrain is most suitable for our experiments. We used the E. coli substrains MG1655 [4], TOP10 (Invitrogen), NEB Turbo (NEB), BAP1 [5] and Rosetta (Novagen). In order to optimize the yield of indigoidine produced by indC, we investigated the influence of the T-domain on the enzyme efficiency. Previous studies showed, that exchanging the T-domain of the blue pigment synthetase bpsA from S. lavendulae [6] yields an unfunctional indigoidine synthetase [7].We replaced the T-domain of the wild-type indC with T-domains from several other NRPS modules from different host organisms (S. lavendulae, E. coli, B. parabrevis, D. acidovorans, P. luminescens) observing the change in enzyme efficiency (Fig. 2a) [2][3][6][8]. Moreover, we created seven fully synthetic T-domains based on multiple sequence alignments of 250 NRPS modules and measured the indigoidine yield after insertion into the indigoidine synthetase scaffold. Since the the NRPS T-domains must be activated by a member of the 4'-Phosphopantheteinyl-transferases (PPTases) [9], which differ in their substrate specificity [10], we coexpressed every engineered indigoidine synthetase with PPTases from different host organisms (Sfp from B. subtilis [11], Svp from S. verticillus [12], EntD from E. coli [7], DelC from D. acidovorans [8], NgrA from P. luminescens [14]) (Fig. 2b) and determined the indigoidine production rate of every engineered indigoidine synthetase in combination with a respective PPTase.
With this approach, we learn more about the interaction of T-domains and PPTases and their influence on the NRPS efficiency. Since this combinatorial approach requires the cloning of large plasmid libraries and hundreds of cotransformations, we established HiCT: High-throughput protocols for circular polymerase extension Cloning and Transformation (please find more information on HiCT on our RFC99 Page) based on CPEC assembly [11]. Moreover, we modeled the indigoidine production of our constructs (please find more detailed information on our Indigoidine Production Model Page)
Results
Expression of Indigoidine Synthetase IndC in Five Substrains of E. coli
The open reading frame of the native indigoidine synthetase indC was amplified from genomic DNA of P. lumninescens laumondii TT01 (DSM15139) and cloned into a pSB1C3 derived plasmid under the control of an lac-inducible promoter. This indC expression cassette was transformed into different substrains of E. coli, namely DH5alpha, MG1655, BAP1, TOP10 and NEB Turbo. All of these host strains were previously transformed with a pSB3K3 derived expression plasmid coding for the PPTase Sfp from B. subtilis, which is commonly used in NRPS studies. As depicted in Fig. 3a, Sfp is able to activate the T-domain of indC as determined by the blue phenotype of the transformed cells. Except for NEB Turbo cells, all transformed host strains displayed a decellerated growth and significantly smaller colonies on plate when compared to the E. coli TOP10 negative control. The blue phenotype developed late after transformation ranging from first blue colonies after 24 h and taking up to three days for visible poduction of the blue pigment. NEB Turbo showed regular colony growth and developed a strong blue phenotype upon induction with IPTG. As all host strains were able to express the functional indigoidine sythetase IndC, further experiments were only conducted with one E. coli strain. Due to its simplicity in handling and sufficient expression of the constructs, the substrain TOP10 was chosen.

Indigoidine Production Varies when Co-expressed with Different PPTases
The expression of indC under activation by the endogenous PPTase entD in E. coli was sufficient for easy detection of indigoidine production on plates Fig. 3b, indC). In order to determine, whether the amount of indigoidine in the E. coli TOP10 cells is dependent on the quality of the interacting of indC with the PPTase, four PPTase dervied from varying origins were selected and amplified from the genome of the hosts of origin. E. coliTOP10 cells were co-transformed with plasmids coding for the different PPTases and the plasmid containing the expression cassette for indC. As reference for the endogenous PPTase activity served cells only transformed with the indC plasmid. Irrespective of the PPTase, growth of colonies was retarded. Remarkably however, colonies co-transformed with the PPTase plasmid remained of smaller size than the ones only carrying the indC construct. On the other side, indigoidine production was more diffuse in the latter cells with secretion of the blue pigment into the agar (Fig. 3b, indC) and only slight blue-greenish coloring of the colonies. The four PPTases additionally introduced into the TOP10 cells were all shown to be functional (blue phenotype of the transformants, Fig. 3b ), but lead to the retention of most of the indigoidine within the cells. Colonies of cells transformed with thess constructs, were of convex shape and of distinct, dark blue color. Overall, cells carrying an additional PPTase showed increased indigoidine production compared to the cells relying on the endogenous entD.
T-Domain Exchanges in IndC Yield Functional Indigoidine Synthetases
The main structural characteristic of NRPSs is their modular composition on different hierarchical levels. The indogoidine synthetase indC is a single module NRPS comprised of the three domains, namely AOxA, T and TE. Since the functionality of this NRPS is detectable by the bare eye, it offers a perfect and simple experimental set-up for proof of principle experiments regarding the interchangeability of domains from different NRPS. Out of the three domains in indC, the T-domain is supposed to exhibit the least substrate specificity and was thus chosen for first domain shuffling approaches. For the initial definition of T-domain boundaries of indC, we used Pfam, a web-tool which allows -amongst other functions- for the prediction of NRPS module and domain boundaries (Pfam.sanger.ac.uk. Following the boundary prediction, we choose a two-pronged domain shuffling approach: First, we transferred native T-domains derived from either different host species and/or NRPS of entirely different function into the indC indigoidine synthetase (Streptomyces lavendulae lavendulae ATCC11924 (blue pigment synthetase bpsA) [6], Brevibacillus parabrevis (Tyrocidine synthesis cluster) [2], Delftia acidovorans SPH-1 (Delftibactin synthesis cluster) [8], Photorhabdus luminescens laumondii TT01 (plu2670 and plu2642, unknown function) [14] and Escherichia coli MG1655 (entF from enterobactin synthesis cluster) [4]). Second, we deviced three methods for the generation of synthetic T-domains based on different NRPS libraries generated by BLAST search against either specific subranges of host organisms or restricting the query sequence to be BLASTed (BLAST.ncbi.nlm.nih.gov). The engineered indigoidine synthetases were coexpressed with supplementary PPTases (sfp, svp, entD and delC) in E. coli TOP10 cells (Fig. 4a).
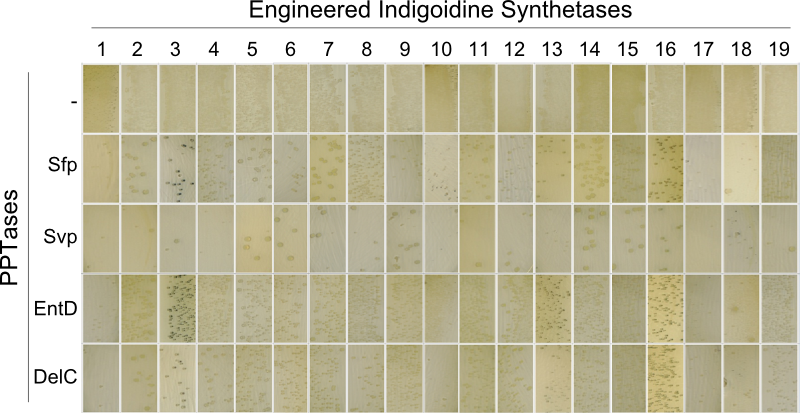
As depicted in Fig. 5, both approaches lead principally to fully functional indCs. The synthetic T-domains 1, 3 and 4 showed the same decreased growth and indigoidine production on plates as did the native T-domain derived from P. lumninescens. The colonies obtained after co-transformation with supplementary PPTase plasmid were small in size and of dark blue color. Compared to synthetic T-domain 5, indigoidine production started earlier (approximately after 24-30 hours).

In contrast to the synthetic domains 1,3 and 4 which were designed by the consensus method and showed medium to high similarity to the sequence of origin, synthetic domain 5 was generated by the guided random method. Remarkably, even though 39 out of the 62 amino acids of the original T-domain were exchanged, the indigoidine synthetase with this T-domain was still functional. Closer analysis of the sequence compared to the original indC T-domain sequence showed, that the characteristics of the amino acid sequence, i. e. for instance polar or charged amino acids, were retained in 72 % of the sequence. Also, the GGxS core sequence of the T-domain at which the activation by the PPTas occurs was conserved.
The Efficiency of Engineered NRPS is Improved When Optimal Domain Borders Are Applied
Multiple web-tools exist which offer the prediction of NRPS module and domain boundaries. One of the most common used prediction tools is Pfam which we used as a starting point to determine the best method for defining domain boundaries. Pfam predicted large linker structures between the end of the A- and the beginning of the T-domain (compare Fig. 5, T-boundaries from "B" to "2"). Using these domain boundaries for the native T-domains did only yield one functional native T-domain (Fig. 5, plu2642). We tried to improve this yield by defining new T-domain boundaries based on the predictions of Pfam and multiple sequence alignments (MSA) with the respective homology libraries at the predicted linker regions. Boundaries were set closer to the preceeding A-domain, at regions were less sequence conservation was observed. Fig. 6 shows the indigoidine production after insertion of native T-domains with revised boundaries. T-domain boundary combinations A1, A2 and C1 yielded functional T-domains. As the indigoidine production and cell growth was best for the T-domains created with boundary combination A2, this boundary design was used for all subsequent cloning strategies.

Fig. 7 depicts the success of this boundary desgin as two additional native T-domains derived from delH4 and bpsA (indigoidine synthetase) led to functional indCs and the production of indigoidine. In addition, the native T-domain from plu2642 which was already shown to be functional (compare Fig. 3b ) showed faster and increased indigoidine production (deep blue agar plate, lower right panel on Fig. 7 ). The results obtained from this experiments proofed two concepts: First, domain shuffling is possible across different species as the T-domains of delH4 and bpsA were derived from D. acidovorans and S. lavendulae lavendulae, respectively and were functional in E. coli. Also, shuffling of domains from modules of different substrate specificity has been proofen herein. Second, manually adjusting the boundaries predicted by Pfam based on MSA is a functional method to predict functional T-domain boundaries.

PPTase and T-domain Interaction Strongly Influence the Yield of Indigoidine Production
As the previous experiments of shuffled T-domains and different combinations of PPTases showed, there are substantial differences in cell growth and indigoidine production when observed on plates. However, this observations were always of qualitative nature and did not give any insight into quantitative differences. We approached the quantification of indigoidine production in a time-resolved and highly-combinatorial manner: plasmids coding for indC containing all synthetic (4) and native T-domains (3) proven functional by the previous assays were co-transformed with the four functional PPTases. The indigoidine production over time (30 hours) was measured at its absorption maximum of 590 nm and corrected for the contribution of the cellular components in the medium as described in the methods. As Fig. 8 shows, synthetic T-domains in combination with different PPTases lead to distinct differneces in indogoidin production.
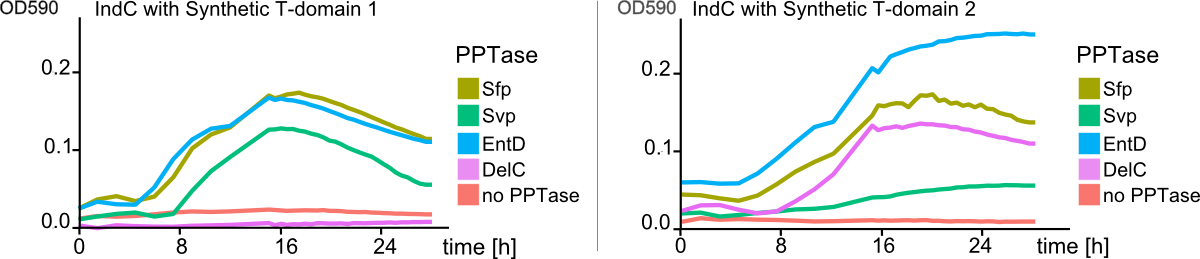
As a comparison between the left and right panel of Fig. 8 shows, PPTases working best with one T-domain might not lead to any indigoidine production when used with a indigoidine synthetase containing a different T-domain (Fig. 8 , pink line, delC). In addition, as distinctly visible in Fig. 9 , indigoidine production over time is not a strictly monoton function (blue line). After an indigoidin production peak at 16 hours, indigoidine production caused by indC containing the synthetic domain 4 decreases again. The indigoidin production in cells transformed with indC/synthetic T-domain 3 is still increasing.

Engineered Indigoidine Synthetase is More Efficient than Native Enzyme
After applying the domain border combination A2 (see Fig. 6
) to all T-domains we inserted to the indC gene, we quantified the amount of indigoidine produced per cell density of E. coli cells expressing both the engineered indC variant and a PPTase (Sfp, Svp, EntD or DelC). We used a Tecan infinite M200 plate reader and applied the quantitative indigoidine assay described in the methods section. Fig. 10
shows the relative indigoidine production of representative samples in this measurement, illustrated as a heat map. The indigidine production strongly varies among different combinations of T-domains and PPTases. Notably, one of the engineered indigoidine synthetases is more efficient than the native enzyme itself. This most efficient indigoidine synthetase carries the synthetic T-domain #3. With this remarkable finding we wanted to have a closer view at structural aspects of this enzyme. Currently, the crystal structures of both the native IndC and our optimized variant are investigated by the group of Dr. Bange (Philipps-Universität Marburg).

Discussion
Motivated by the establishment of the Indigoidine-Tag, which enables the labelling of nonribosomal peptides, we wanted to further investigate the indigoidine synthetase indC [3] and thus the overall NRPS modularity in order to optimize the functionality of the indigoidine tag. In previous studies, direct T-domain exchanges have not been successful in the context of the indigoidine synthetase BpsA from S. lavendulae [6][7]. Furthermore, the endogenous PPTase EntD of E. coli was considered inefficient in the activation of the blue pigment synthetase BpsA [6]. Therefore, most studies in the field of NRPS are conducted with the PPTase Sfp from B. subtilis [11]. The family of 4'-Phosphopantheteinyl-transferases was reported to vary in substrate specificity [11]. Therefore, they can only activate specific NRPS module families [9]. We first investigated the production of indigoidine in five substrains of E. coli: TOP10 (Invitrogen), MG1655 [4], NEB Turbo (NEB), BAP1 [5] and Rosetta (Novagen), using an expression plasmid coding for the indigoidine synthetase IndC and the PPTase Sfp. Though differring in their growth behaviour, all substrains showed to be capable of producing the blue pigment indigoidine (Fig. 3a).
In our approach, we replaced single domains of the indC indigoidine synthetase module with both respective domains of other NRPS pathways from different organisms and entirely synthetic domains which were created based on multiple sequence alignments of 250 NRPS domains. We also created indC variants, in which different domain borders have been applied for the domain exchange (Fig. 6), since setting the correct linker sequence is crucial for the NRPS function [7]. We used NRPS domains from Streptomyces lavendulae lavendulae ATCC11924 (blue pigment synthetase bpsA) [6], Brevibacillus parabrevis (Tyrocidine synthesis cluster) [2], Delftia acidovorans SPH-1 (Delftibactin synthesis cluster) [8], Photorhabdus luminescens laumondii TT01 (plu2670 and plu2642, unknown function) [14] and Escherichia coli MG1655 (entF from enterobactin synthesis cluster) [4], thus creating a library of 58 different engineered indigoidine synthetases. We co-transformed the 58 indC variants with four different PPTases into E. coli TOP10 cells. We used the PPTases Sfp (Bacillus subtilis str. 168) [11], Svp (Streptomyces verticillus ATCC15003) [12], EntD (Escherichia coli MG1655) [7] and DelC (Delftia acidovorans SPH-1) [8] and screened them on their functionality. This screening is remarkably easy, because functional indigoidine synthetases result in a blue phenotype when being expressed and activated in E. coli cells [3]. We found, that nine engineered indigoidine synthetases, in which the T-domain was replaced, remained functional in producing indigoidine when co-expressed with specific PPTases. The inserted T-domains include four synthetic T-domains (Fig. 4
), the T-domain of bpsA with three different domain border combinations (Fig. 6) and the T-domains of the NRPS modules plu2642 (P. luminescens) and delH (D. acidovorans)(Fig. 7). In order to quantify the amount of indigoidine produced by the engineered indigoidine synthetases when co-transformed with different PPTases, we established a quantitative indigoidine assay based on OD measurement using a Tecan infinite M200 plate reader, inspired by Myers et al. [15]. Notably, some of our engineered IndC constructs were more efficient in the production of indigoidine compared to the wild-type IndC (T-domains synT1, synT3, synT4; Fig. 10).
This is particularly remarkable as our results contradict to previous studies of NRPS domains that reported the native T-domain of the indigoidine synthetase BpsA to be essential for protein function (and therefore not replaceable by other T-domains)[7].
In conclusion, we were able to demonstrate that it is indeed possible to replace single domains from NRPS modules, while preserving or even enhancing their functionality. In addition, we established an approach for the design of synthetic T-domains and proved their functionality by introducing them into the indigoidine synthetase indC scaffold. Moreover, we established a high throughput protocol for circular polymerase extension cloning and transformation (Hi-CT) (BBF RFC 99) based on CPEC assembly [13], which we applied for our domain shuffling approach. In summary, we created a library of 58 engineered indC variants. In addition we perforemd measurement of blue pigement production over time, which gave us novel insights in how NRPS domains should be designed, where the domain borders between different domains in a single NRPS module have to be set and which domains from respective NRPS pathways and bacterial strains can be used, when creating novel engineered NRPS pathways. We implemented our findings into the "NRPS-Designer" Software, so that the underlying algorithm for NRPS design takes into consideration the abovementioned findings (e.g. domain border setting) which are certainly crucial for successful in silico prediction of functional NRPSs. Moreover, we modeled the indigoidine production of our constructs (please find more detailed information on our Indigoidine Production Model Page). The crystal structure of our optimized indigoidine synthetase, which is currently investigated by the group of Dr. Bange (Philipps-University Marburg), will give more insight into structural aspects of non-ribosomal peptide synthesis.
Thereby, our project pioneers the research on high-throughput methods for creation and optimization of synthetic NRPS modules composed of user-defined domains. We believe that our findings will highly contribute to future development of custom NRPSs.
Methods
Cloning Strategy
We assembled the different indC variants on a chloramphenicol resistance backbone (pSB1C3) with an IPTG-inducable lac-promoter, the ribosome binding site BBa_B0034 and the coding sequence of the respective indC variant. The indC plasmids should be co-transformed with a PPTase construct to get a significant and fast indigoidine production. Therefore, we used a second plasmid backbone carrying a kanamycin resistance (pSB3K3). We assembled five pSB3K3 derived plasmids, each carrying an expression cassette with an IPTG induceable lac-promotor, the BBa_B0029 ribosome binding site and the coding sequence of the respective PPTase (sfp, svp, entD, delC and ngrA). We used E. coli TOP10 for co-transformations of the possible combination of the indC variants and all PPTase plasmids.Circular Polymerase Extension Cloning
Circular Polymerase Extension Cloning (CPEC) is a sequence-independent cloning method based on homologous recombination of double-strand DNA overlaps of vector and insert(s) (Fig. 11)(Quan 2008). It is suitable for the generation of combinatorial, synthetic construct libraries as it allows for multi-fragment assembly in an accurate, efficient and economical manner.
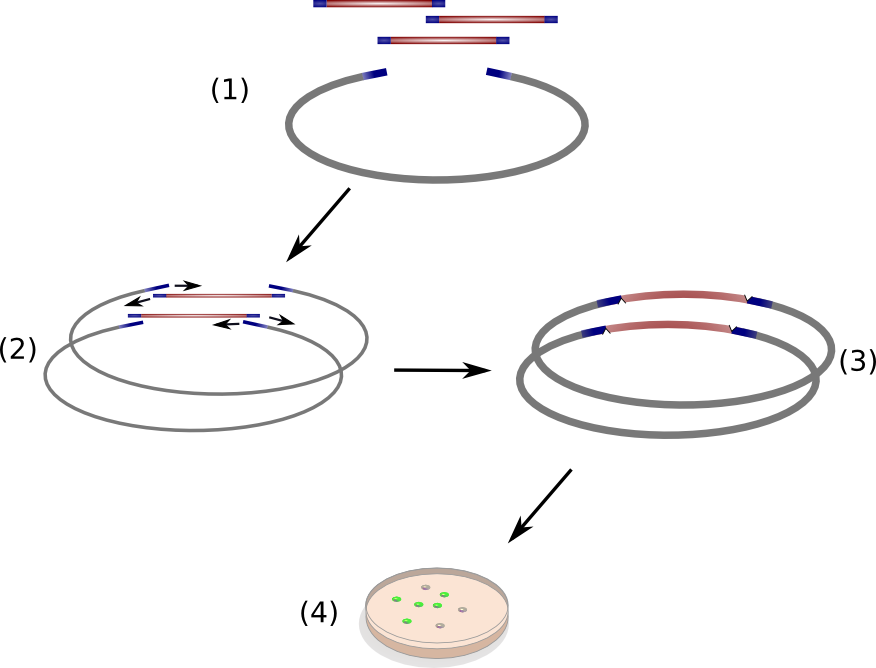
CPEC relies on a simple polymerase extension of the DNA fragments to be assembled. Crucial to this concept is the design of vector and insert fragments which must share overlapping regions at the ends (Fig. 11 (1)
). In a single reaction set-up, insert DNA fragments and linear vector are heat denaturized and allowed to anneal at elevated temperature, resulting in specific hybridized insert-vector constructs (Fig. 11 (2)
). Subsequently, the single-strand hybrid constructs are extended under PCR-elongation conditions (72 °C for 20 s/kbp of longest fragment) which yield completely assembled, double-stranded circular constructs (Fig. 11 (3)
) ready for transformation into competent cells. The single strands nicks introduced on each strand due to the unidirectional nature of the polymerase chain reaction will be removed by endogenous ligases upon transformation into Escherichia coli.
We provide instructions (RFC 99) for a rapid and cost efficient cloning and transformation method based on CPEC which allows for the manufacturing of multi-fragment plasmid constructs in a parallelized manner: High Throughput Circular Extension Cloning and Transformation (HiCT)
CPEC was performed according to the following protocol: The total mass of DNA used per CPEC reaction varied between 50 to 200 ng. The insert to backbone molar ratio was 3:1 for insert-backbone and 1:1 for insert-insert molar ratio. Conversion from mass concentration of fragments to molar concentration was done using the formula: cM = c*10^6/(n*660), where c is the measured oligonucleotide concentration [ng/µl], n is the number of dinucleotides of the fragment and cM is the resulting concentration [nM]. The final reaction volume was adjusted to 6 µl with polymerase master mix (Phusion® High-Fidelity PCR Master Mix with HF Buffer, NEB #M0531S/L). The CPEC reaction was carried out under the following conditions:
- initial denaturation at 98°C for 30 s
- 5 cycles with:
- denaturation step at 98°C for 5 s.
- annealing step at 53°C for 15 s
- elongation/filling up step at 72°C for 20 s/kbp of longest fragment.
- final extension at 72°C for three times the calculated elongation time.
- (Optional: Hold at 12°C )
After CPEC, 5 µl of of the reaction mixture were used for transformation. The remaining volume was used for quality check on a gel with small pockets (10 to 20 µl in volume).
Generation of cdB-indC Construct
To minimize the background colonies when exchanging the T-domain of the indigoidine synthetase we generated the ccdB-Ind plasmid where we replaced the indC T-domain with the ccdB gene (Modul structure: AoxA-ccdb-TE) which kills E. coli TOP10 cells but not E. coli OneShot ccdB survival cells. Test-transformation in both E. coli TOP10 and the E. coli OneShot ccdB survival cells showed that background colonies could be eliminated by this strategy. We used the ccdB-Ind for all further CPEC experiments aiming to swap T-domains. Primers for the backbone CPEC fragments were designed to facilitate the amplification of the entire ccdB-Ind plasmid while omitting the ccdB sequence. Assembly of the finale indigoidine synthase products with exchanged T-domain was achieved by CPEC as described or above or HiCT (RFC 99).
Examination of T-domain Borders
We exchanged the T-domain of indC with the T-domain of bpsA and varied the size of the exchanged DNA sequence, thus examining several domain borders (Figure 5a). We used the CPEC assembly method and the indC-ccdB plasmid for this approach. For the investigation of additional T-domains from less related NRPS modules, we selected the border combination A2 which was positive in the test with bpsA. We used the T-domains of the following genes:
| Gene | Organism | Original function |
|---|---|---|
| entF | Escherichia coli K-12 | NRPS module of enterobactin synthesis pathway |
| tycA1 | Brevibacillus parabrevis | 1st module in tyrocidine synthesis cluster |
| tycC6 | Brevibacillus parabrevis | Last module in tyrocidine synthesis cluster |
| delH4 | Delftia acidovorans SPH-1 | 2nd but last module in delftibactin synthesis cluster |
| delH5 | Delftia acidovorans SPH-1 | Last module in delftibaction synthesis cluster |
| plu2642 | P. luminescens DSM15139 | NRPS of unknown function (one module: A-T-TE) |
| plu2670 | P. luminescens DSM15139 | module of NRPS pathway of unknown function |
All T-domains from the respective genomes were amplified using CPEC primers with a uniform 5’-end and a 3’-end specific for the respective gene. For the assembly of the hybrid-indigoidine synthetases by CPEC, the indC-ccdB construct was used.
Creation of Synthetic T-domains
All R scripts used in the following sections are based on R version R-3.0.1. Different assumptions about the evolutionary conservation of T-domains were examined: i) conservation of a specific module across different species, ii) conservation of T-domains across different modules for the same species, iii) conservation of T-domains across different species, iv) conservation of similar modules across different species. According to these three assumptions, different libraries of homologous protein sequences were generated using ncbi protein BLAST (blast.ncbi.nlm.nih.gov) with standard parameters:
- query sequence: indC; Search set: non-redundant protein sequences without organism restriction
- query sequence: indC T-domain; Search set: non-redundant protein sequences within P. luminescens
- query sequence: indC T-domain; Search set: non-redundant protein sequences without organism restriction
- query sequences: indC, bpsA, entF, delH5 and tycC6; Search set: non-redundant protein sequences without organism restriction;
The 50 closest related protein sequences contained in each the library were subjected to a multiple sequence alignment (MSA) using clustalO (http://www.ebi.ac.uk/Tools/msa/clustalo/). with standard parameters for protein alignments. For library generation iv), each query sequence was BLASTed separately and the 50 best results of each query were combined i.e. a total of 250 sequences for the MSA. After library generation, the following three methods were employed to design different synthetic T-domains.
1. Consensus Method
Based on the .clustal file obtained from the MSA of the homology libraries, a consensus sequence using the UGENE software (http://ugene.unipro.ru/) with a threshold of 50% was created (i.e. if an amino acid appears in 50% or more of all sequences at a specific position it is considered as a consensus amino acid). For the creation of the synthetic T-domains, this consensus sequence was used to fill the gaps where there was no consensus amino acid with the original amino acid from the indC T-domain. By this approach, T-domains were generated which might deviate from the original sequence at positions with at least average conservation but coreespond to the original one if there is less conservation.
2. Guided Random Method
In this approach, the multiple sequence alignments (MSA) generated by the consensus method was used. Implemented in R, a position-specific profile was generated which has the same length as the MSA and contains the rate at which amino acids occur at any given position of the sequence alignment. The synthetic T-domain is created by position-wise generation of the sequence where the probability of choosing an amino acid at a given position is determined by the rate in the profile.
3. Randomized Generation Method
For generation of synthetic sequences by the randomized generation method, every amino acid was assigned a score of 1 or 0, i.e. occuring at least ones or not at all at a given position in the MSA. In the subsequent generation of the synthetic T-domain sequence of the synthetic domain, any amino acid assigned 1 had the same likelihood of being chosen at this position. Seven synthetic T-domains were designed based on differnt combinations of the homology libraries and sequence generation methods.
| Domain ID | Homology library | Sequence generation method |
|---|---|---|
| synT1 | library i | consensus |
| synT2 | library ii | consensus |
| synT3 | library iii | consensus |
| synT4 | library iv | consensus |
| synT5 | library i | guided random |
| synT6 | library iv | guided random |
| synT7 | library i | randomized generation |
Fig. 12 shows the multiple sequence alignment of the seven synthetic T-domains and the native indC T-domain. After the generation of the T-domain amino acid sequences, the OPTIMIZER web-tool(http://genomes.urv.es/OPTIMIZER/) was used to obtain the corresponding DNA sequence [16]. E. coli K-12 was set as strain for codon optimization and most frequent was chosen as codon option. The generated DNA sequence was cured from internal RFC10 cutting sites and CPEC cloning overhang required for the T-domain swapping into the ccdb construct were introduced. The synthetic T-domains were ordered at IDT (Integrated DNA Technologies, Coralville, Iowa). In order to obtain sufficient amounts of DNA, the synthetic T-domains were amplified via PCR. IndC-hybrid constructs of the native IndC with exchange of the native T-domain by the synthetic variants were assembled using CPEC and the indC-ccdB construct as backbone. The synthetic T-domains were amplified for CPEC using the same primers as for the native indC T-domain.

Quantitative Indigoidine Production Assay
1. OD Spectrum Measurement
96-well plates are prepared with 100 µl LB-medium/well containing appropriate antibiotics (chloramphenicol and kanamycin for the indigoidine and PPTase contrcuts, respectively) and each well is inoculated with single colonies (in duplicates) from plates positive for the co-tansformation experiments i.e. from plates with blue colonies. Two sets of negative controls are also inoculated on the plate: First, pure medium serving as the baseline for background correction for the OD measurements. Second, transformation controls accounting for potential differences in cell growth due to expression of proteins contained on the plasmids, i.e. the antibitotic resistance gene and IndC. In this set of controls, the plasmid used in co-transformation with the PPTase plasmid contains IndC-constructs carrying a randomly generated sequence instead of the T-domain. A second 96 well plate was prepared with 180 µl LB-medium/well for the measurement itself. The 96-well plate containing the pre-cultures of the co-transformed colonies was inoculated for 24 hours at 37°C. Subsequently, 20 µl of the pre-culture was transferred to the measurement plate. The absorbance of the bacterial cultures was measured at wavelengths ranging from 400 nm to 800 nm in intervals of 10 nm for each well every 30 min for 30 hours at 30°C in a Tecan infinite M200 plate reader. For the measurement plate, Greiner 96-well flat black plates with a clear lid were used.
2. Data Analysis
Detecting the amount of the NRP expressed by the bacterial host strain is desirable. By tagging the NRP with indigoidine, the amount of the fusion peptide can be determined by quantifying the amount of blue pigment present in the cells. As the amount of blue pigment is proportional to the amount of the NRP of interest, a method for the quantification of the blue pigment will yield information about the expression of the NRP. Quantification of the pure indigoidine pigment can be easily achieved by optical density (OD) measurements at its maximum wavelength of about 590 nm. In cellular culture, indigoidine quantification by OD measurements is impaired. Cellular density of liquid cultures is standardly measured as the optical density (OD) at a wave length of 600 nm, i. e. the absorption peak of indigoidine interferes with the measurement of cell density at the preferred wave length (compare to Fig. 13, grey dashed line). Thus, for measurement of NRP expression without time consuming a priori purification of the tagged-protein, a method to separate the cellular and pigment-derived contributions to the OD is required (compare to Fig. 13, brown and blue lines, respectively).

The method of choice, as described by Myers et al. [15], requires the OD measurement of cell culture at two distinct wavelengths: the robust wave length ODR and the sensitive wave length ODS. The concentration of indigoidine will have to be deducted from measurements at ODS = 590 nm: $$OD_{S,+P}$$ $$[Indigoidine]= OD_{S,+P}-OD_{S,-P}$$ with $OD_{S,+P}$ being the overall OD measurement and $OD_{S,-P}$ being the scattering contribution of the cellular components at the sensitive OD. The scattering contribution of the cellular compenents at $OD_S$ ($OD_{S,-P}$) can be calculated from the scattering contribution measured at the robust wave length according to the following formula: $OD_{S,-P}= \delta*OD_R$. The correction factor $\delta$ is be determined by measuring the OD of pure cellular culture without indigoidine at both the wavelength $[OD]_{S,-P}$ and $[OD]_R$ and calculating their ratio. Finally, the indigoidine production can be determined as $$[Indigoidine]=OD_{S,+P}- \delta*OD_{R}$$ For the calculation of the cellular component when measuring indigoidine producing liquid cell cultures, OD measurement at 800 nm as robust wavelength is recommended. By the approach described above, quantitative observation of the indigoidine production in a liquid culture over time as well as the indigoidine production in relation to the cell growth can be conducted. Background correction i. e. the contribution of the culture medium to the OD measurement is achieved by subtracting the mean of pure culture medium replicates from all OD values measured (Fig. 14).

1. Marahiel MA (2009) Working outside the protein-synthesis rules: insights into non-ribosomal peptide synthesis. J Pept Sci 15: 799-807.
2. Doekel S, Marahiel MA (2000) Dipeptide formation on engineered hybrid peptide synthetases. Chem Biol 7: 373-384.
3. Brachmann AO, Bode HB (2013) Identification and Bioanalysis of Natural Products from Insect Symbionts and Pathogens. Adv Biochem Eng Biotechnol
4. Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y (1997) The complete genome sequence of Escherichia coli K-12. Science 277: 1453-1462.
5. Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C (2001) Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 291: 1790-1792.
6. Takahashi H, Kumagai T, Kitani K, Mori M, Matoba Y, Sugiyama M (2007) Cloning and characterization of a Streptomyces single module type non-ribosomal peptide synthetase catalyzing a blue pigment synthesis. J Biol Chem 282: 9073-9081.
7. Owen JG, Robins KJ, Parachin NS, Ackerley DF (2012) A functional screen for recovery of 4'-phosphopantetheinyl transferase and associated natural product biosynthesis genes from metagenome libraries. Environ Microbiol 14: 1198-1209.
8. Johnston CW, Wyatt MA, Li X, Ibrahim A, Shuster J, Southam G, Magarvey NA (2013) Gold biomineralization by a metallophore from a gold-associated microbe. Nat Chem Biol 9: 241-243.
9. Fischbach MA, Walsh CT (2006) Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev 106: 3468-3496.
10. Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, Reid R, Khosla C, Walsh CT (1996) A new enzyme superfamily - the phosphopantetheinyl transferases. Chem Biol 3: 923-936.
11. Nakano MM, Corbell N, Besson J, Zuber P (1992) Isolation and characterization of sfp: a gene that functions in the production of the lipopeptide biosurfactant, surfactin, in Bacillus subtilis. Mol Gen Genet 232: 313-321.
12. Sanchez C, Du L, Edwards DJ, Toney MD, Shen B (2001) Cloning and characterization of a phosphopantetheinyl transferase from Streptomyces verticillus ATCC15003, the producer of the hybrid peptide-polyketide antitumor drug bleomycin. Chem Biol 8: 725-738.
13. Quan J, Tian J (2009) Circular polymerase extension cloning of complex gene libraries and pathways. PLoS One 4: e6441.
14. Duchaud E, Rusniok C, Frangeul L, Buchrieser C, Givaudan A, Taourit S, Bocs S, Boursaux-Eude C, Chandler M, Charles JF, Dassa E, Derose R, Derzelle S, Freyssinet G, Gaudriault S, Medigue C, Lanois A, Powell K, Siguier P, Vincent R, Wingate V, Zouine M, Glaser P, Boemare N, Danchin A, Kunst F (2003) The genome sequence of the entomopathogenic bacterium Photorhabdus luminescens. Nat Biotechnol 21: 1307-1313.
15. Myers JA, Curtis BS, Curtis WR (2013) Improving accuracy of cell and chromophore concentration measurements using optical density. Bmc Biophysics 6:
16. Puigbo P, Guzman E, Romeu A, Garcia-Vallve S (2007) OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res 35: W126-131.
