 "
"
Team:Imperial College/Waste Degradation: SRF
From 2013.igem.org





Module 1: Resource-full Waste

Non-recyclable waste is sourced from a recycling centre, placed in a bioreactor with our M.A.P.L.E system which degrades the waste and synthesises the bioplastic P(3HB). Click the tabs to find out more
Overview
Specifications
Design
Modelling
Assembly
Testing & results
Future work
Waste from recovery facilities is a mixture of plastics and cellulosic rich biomass such as fibres and wood. The Resource-full waste module utilises the large variety of materials organisms can naturally degrade to recycle the waste and to provide food for the production of the bioplastic poly-3-hydroxybutyrate, P(3HB).
Separating polyurethane and selling its breakdown products
Certain enzymes have the ability to degrade plastics as a result of their evolutionary history, dealing with tough and highly variable substrates such as lignin. Various enzymes have been biobricked to deal with most of the major petrochemical plastics, except for polyurethane. Polyurethane is one of the plastic constituents of this mixed waste. We have designed biobricks containing naturally occurring polyurethane esterases (also known as PUR esterases) to break polyurethane down into its constituent chemicals of ethylene glycol and poly(isocyanate). These valuable chemicals will be separated using specialised filters and sold back to industry.
Using the residual material as a substrate for P(3HB) production
The remaining organic portion of the mixed waste is used as a carbon source for P(3HB) production. P(3HB) is produced by organisms containing the pha CAB operon, originally found in soil bacteria such as Ralstonia eutropha. The operon contains three genes which are members of the pathway required to make P(3HB). P(3HB) is used as a storage molecule in the organisms which produce it. They do this when nutrients are limiting but when there is a plentiful carbon source.
P(3HB) is produced commercially by several companies. One of the crucial factors in whether production is economical or not is the efficiency of bioplastic production. We have hugely increased the efficiency of P(3HB) production; an important step in making our system feasible.
To scale up the production of P(3HB), our system would require large amounts of purified sugars. We would, therefore, hydrolyse the long polysaccharide components to release the sugars. This is a current industrial pre-treatment technique to allow the fermentation of ligno-cellulosic biomass.

References:
- Harding KG, Dennis JS, von Blottnitz H, Harrison STL. Environmental analysis of plastic production processes: Comparing petroleum-based polypropylene and polyethylene with biologically-based poly-beta-hydroxybutyric acid using life cycle analysis. J Biotechnol 2007 MAY 31;130(1):57-66.
- Kim S, Dale BE. Energy and Greenhouse Gas Profiles of Polyhydroxybutyrates Derived from Corn Grain: A Life Cycle Perspective. Environ Sci Technol 2008 OCT 15;42(20):7690-7695.
- Kendall A. A life cycle assessment of biopolymer production from material recovery facility residuals. Resources, Conservation and Recycling 61 (2012) 69– 74 conomou A. Following the leader: bacterial protein export through the Sec pathway. Trends in microbiology 1999;7(8) 315-320.
- Chul-Hyung Kang, Ki-Hoon Oh, Mi-Hwa Lee, Tae-Kwang Oh, Bong Hee Kim, Jung- Hoon Yoon. A novel family VII esterase with industrial potential from compost metagenomic libraryMicrobial Cell Factories 2011, 10:41 doi:10.1186/1475-2859-10-41
- Da Almeida, Alejandra. Effect of the granule associated protein phasin (PhaP) on cell growth and poly(3-hydroxybutyrate) (PHB) accumulation from glycerol in bioreactor cultures of recombinant E. coli. Volume 131, Issue 2, Supplement, September 2007, Pages S167.
Specification
1. Our bacteria should survive and grow in mixed waste
In order for the bacteria to produce bioplastic from the mixed waste they first need to be able to use it as a carbon source
2. Our bacteria should secrete a functional polyurethane esterase
To recover the resources from polyurethane in the mixed waste, the enzymes should secrete in an active form.
3. Our bacteria should be able to tolerate mixed waste degradation products, such as ethylene glycol
The products of polyurethane degradation are toxic and so any bacteria growing with them must be able to survive to a concentration which will allow economical production of ethylene glycol. We have chosen a strain of E.coli-MG1655, which is resistant to ethylene glycol toxicity.
4. Our bacteria should produce P(3HB)
5. Our bacteria should produce P(3HB) from the mixed waste
PUR Degradation: Pathway ▼

Lignin degrading microbial enzymes are capable of degrading plastics. Some previous iGEM teams have exploited this ability. We have built on their work and extended the plastic degradation capabilities of the synthetic biology community by improving polyurethane (PUR) degradation, as this has not been successfully achieved before. We have identified 5 PUR-esterase enzymes from the literature that are capable of catalyzing the below reaction.

We have synthesised all of the genes in the below table and are testing them for expression in E.coli, secretion, activity and PUR degradation capabilities. Our ultimate design is to be able to control the relative levels of different enzymes in a waste-degrading bio-reactor, in order to adjust it to the composition of waste. We have, therefore, designed the expression constructs accordingly. Our models predict the degradation rate of polyurethane at the bioreactor scale. .
PUR Degradation: BioBrick Designs ▼
| enzyme | source organism | biobrick | reference |
|---|---|---|---|
| EstCS2 | uncultured unknown bacterium (GU256649.1) | BBa_K1149002 | Kang et.al 2011 |
| pueA | Pseudomonas chlororaphis | BBa_K1149003 | Stern et al., 2000 |
| pueB | Pseudomonas chlororaphis | BBa_K1149004 | Howard et al., 2001 |
| pudA | Comamonas acidovorans | BBa_K1149005 | Allen et al. 1999 |
| pulA | Pseudomonas fluorescens | BBa_K1149006 | Vega et al., 1999 |

Secretion Strategy:
E.coli is commonly used as a chassis in innovative iGEM projects that aim to prove a concept and make the case for a novel function in a biologically engineered machine. In our case, we aim to degrade and synthesise plastic and the degradation part of our system needs to be extracellular. There are many strategies for secretion. You can read about these on our Secretion Guide page and find the corresponding biobricks and projects. If you are currently in the process of designing a Genetically Engineered Machine in E.coli, this page provides a good starting point to look for information.
We chose the pelB secretion tag as it has been demonstrated to work in many cases, sometimes with very high transport efficiency (1)(2). pelB has been used in iGEM projects for many years and is part of 50+ constructs. The UC-Davis team last year used it to secrete LC-Cutinase, a PET plastic degrading enzyme (BBa_K936013) successfully, which is somewhat similar to our plastic-degrading enzymes.
Safety:
We considered the safety aspects of using the PudA enzyme, since its sequences were originally from a Risk Group 2 (RG2) organism. We submitted our safety information to iGEM for review. Safety forms were approved on October 2nd, 2013 by the iGEM Safety Committee. We did not use the organism only the part itself and the final sequences were codon optimised for expression in E. coli, which included the elimination of the forbidden restriction sites.
References
- Thanassi DG, Hultgren SJ. Multiple pathways allow protein secretion across the bacterial outer membrane. Current opinion in cell biology 2000;12(4) 420-430.
- Sletta H, Tondervik A, Hakvag S, Aune TEV, Nedal A, Aune R, et al. The presence of N-terminal secretion signal sequences leads to strong stimulation of the total expression levels of three tested medically important proteins during high-cell-density cultivations of E.coli. Applied and Environmental Microbiology 2007;73(3) 906-912.
Bioplastic Synthesis: Pathway ▼
Poly-3-hydroxybutyrate P(3HB) is a polyester which is naturally produced inside bacteria such as Ralstonia eutropha, where it accumulates as granules inside the cell. In its native bacteria it is produced as an energy store (1).It's properties as a plastic has gained it the attention of researchers and industrialists. In our system, the bacteria will take up organic molecules from waste and use them for P(3HB) production.

We have made P(3HB) in E.coli, transferring three genes naturally found in Ralstonia eutropha into E.coli MG1655. These encode the three enzymes necessary for P(3HB) production; polyhydroxyalkanoate synthase(phaC), 3-ketothiolase(phaA) and acetoacetyl coenzyme A reductase(phaB). These are encoded by the pha CAB operon. We have altered the expression of these three genes to maximise the production of P(3HB) as high yields are required for it to be economically viable.
3-ketothiolase:

Acetoacetyl coenzyme A reductase:

Polyhydroxyalkanoate synthase:

Bioplastic Synthesis: Biobrick Designs ▼
We used the pha CAB biobrick BBa_K934001 to produce P(3HB) which contains the native operon of Ralstonia eutropha. Inthe future we would like to see our system implemented at an industrial scale. This requires high yields to be economically viable. We created P(3HB) synthesis models to inform our experimental attempts to increase P(3HB) yield. We were especially interested to find out if up or down-regulation of any of the enzymes involved in the biosynthetic pathway could increase yields. To analyse this, we needed to know the reactions in the metabolic pathway and also the relative parameters of the enzymes and their dynamic relationships in order to identify bottlenecks in the flux of metabolites. The results from the metabolic model suggest that the amount of pha B is especially critical and increasing its level should give us more P(3HB). (Please see our modelling section for details.) To do this, we designed constructs with stronger promoters to up-regulate the expression of the operon.
We designed and constructed two new constructs for increased expression of the operon (including pha B). The first construct uses the constitutive promoter J23104 and the RBS 0034 with a following scar site TACTAGAG in front of the ATG of the pha C gene. The second construct was designed as a hybrid promoter incorporating BBa_J23104 and the original promoter first, followed by the native promoter and RBS, due to recent results of high expression from a hybrid promoter[1]. The region containing the native promoter and RBS is 352 nucleotides long and might contain important regulatory elements and therefore we were interested to test the hybrid construct in comparison to both the native and constitutive promoter configurations.</

Chassis Design: Tolerance to Waste ▼
We chose the MG1655 E.coli strain as chassis because it constitutively expresses genes that make it resistant to toxic waste degradation products, such as Ethylene glycol. The aldA and fucO genes have an important role in decreasing the toxic effects of ethylene glycol by converting it into glycolaldehyde which is a link to the cell`s central metabolism. We have received these genes from the registry and future work could be to express these in Bacillus or cellulose-degrading organisms to make them tolerant as well.

Enzyme Secretion Strategy ▼
E.coli is commonly used as a chassis in innovative iGEM projects that aim to prove a concept and make the case for a novel function in a biologically engineered machine. In our case, we aim to degrade and synthesise plastic and the degradation part of our system needs to be extracellular. There are many strategies for secretion. You can read about these on our Secretion Guide page and find the corresponding biobricks and projects. If you are currently in the process of designing a Genetically Engineered Machine in E.coli, this page provides a good starting point to look for information.
We chose the pelB secretion tag as it has been demonstrated to work in many cases, sometimes with very high transport efficiency (3). pelB has been used in iGEM projects for many years and is part of 50+ constructs. The UC-Davis team last year used it to secrete LC-Cutinase, a PET plastic degrading enzyme [2] successfully, which is somewhat similar to our plastic-degrading enzymes.
References
- Economou A. Following the leader: bacterial protein export through the Sec pathway. Trends in microbiology 1999;7(8) 315-320.
- Thanassi DG, Hultgren SJ. Multiple pathways allow protein secretion across the bacterial outer membrane. Current opinion in cell biology 2000;12(4) 420-430.
- Sletta H, Tondervik A, Hakvag S, Aune TEV, Nedal A, Aune R, et al. The presence of N-terminal secretion signal sequences leads to strong stimulation of the total expression levels of three tested medically important proteins during high-cell-density cultivations of E.coli. Applied and Environmental Microbiology 2007;73(3) 906-912.
Safety ▼
Safety issues regarding our chassis can be found here.
Modelling
What we learnt from the PUR degradation model:
- The model assisted the design of enzyme assays, in particular:
- The toxicity of the PUR degradation product, Ethylene glycol was taken into account in the modelling and it successfully determined the maximum tolerance of the system.
- Ethylene glycol toxicity assays were carried out in the wet lab and were designed according to the results from our model.
What we learnt from the P(3HB) synthesis model:
- Scanning concentration of pha B and sensitivity analysis showed that increasing the concentration of pha B would increase the production rate of P(3HB) in our engineered 'E.coli'.
- Simulations showed that the constitutive promoter J23104 could lead to a higher expression of pha B than the native promoter. As a result the Wet lab team designed and built BBa_K1149051, which significantly increased bioplastic production.
- Results from the metabolic model suggested that the synthesis of P(3HB) would be accompanied by a drop of ATP over time and hence allowed us to identify potential limitations of our engineered system. This is important for the Industrial Implementation of our system.
1.0 Polyurethane (PUR) degradation model ▼
Introduction
The efficiencies for polyurethane (PUR) degradation and ethylene glycol production are important for the performance of our system. We built a mathematical and deterministic model that is based on MATLAB extension Simbiology for polyurethane degradation. The model contains the kinetic properties of degradation enzymes that are helpful for the design of assays. As we scaled up the initial concentrations of all substrates to meet the conditions for a bio-reactor, the model can provide preliminary simulations and predictions for the MAPLE system.
Design
Objective
Here are some specific objectives for the model to achieve:
1. The model should contain the gene expression model of the degradation enzymes because the enzyme concentration determines the rate of plastic degradation. In our case for PUR degradation, we used pBAD strong promoter K206000 for most enzymes. We built the gene expression model based on inducible pBAD promoter, which gene expression rate can be regulated by inducer concentration.
2. The model should show the efficiency of the enzyme secretion to the culture from the cells. It's also important because the enzyme concentration in the culture depends on it. Here we used pelB secretion tag for most enzymes in order to achieve a high efficiency.
3. The model basically predicts how long will take to degrade a known concentration of soluble polyurethane. It is assumed that the enzyme in our assays has the same kinetic properties as the enzyme used in the literature. The model can suggest a suitable concentration of the plastic to use in order to get good results from the assays.
4. It is known that ethylene glycol is toxic to E.coli. It has, however, no clear effect on the growth of our MG1655 strain when the concentration of ethylene glycol is below 200 mM. The model, therefore, should suggest a safe range of polyurethane concentration to avoid a high concentration (>200mM) of ethylene glycol produced.
The Model
Polyurethane (PUR) degradation involves 5 different degradation enzymes:
| enzyme | source organism | biobrick | reference |
|---|---|---|---|
| EstCS2 | uncultured unknown bacterium (GU256649.1) | BBa_K1149002 | Kang et.al 2011 |
| pueA | Pseudomonas chlororaphis | BBa_K1149003 | Stern et al., 2000 |
| pueB | Pseudomonas chlororaphis | BBa_K1149004 | Howard et al., 2001 |
| pudA | Comamonas acidovorans | BBa_K1149005 | Allen et al. 1999 |
| pulA | Pseudomonas fluorescens | BBa_K1149006 | Vega et al., 1999 |
4 of the 5 enzymes are not well characterised before, so we could't find enough kinetic data from the literature. The only well-characterised PUR degradation enzyme PudA is used in the model as an illustration of all PUR degradation enzymes. The model will be more complete when the kinetic data of the other enzymes are defined. The finished PUR degradation model is shown as below:

There are two compartments which represents cells and the culture from left to right. The "cell" compartment contains the gene expression module whereas the "culture" compartment contains the degradation module. The "secretion" block that connects two compartments is the secretion module.
Parameters and assumptions
| Parameter | Description | Value | Units | Source | Assumptions/Notes |
|---|---|---|---|---|---|
| β | maximum rate of transcription | 0.015 | mM/min | Please see derivation 1 below. | Please see derivation 1 below. |
| K | Activation coefficient | 0.0031 | mM | [3] | Taking the "switch point" as the activation coefficient |
| dmRNA | mRNA degradation rate | 0.035 | 1/min | [4] | There is no active degradation pathway and that dilution is the dominant way by which it degrades. Rate = ln2/doubling time, where doubling time of strain MG1655 = 20min. Assuming steady-state growth in LB broth as presented in paper. rate = ln2/20 = 0.035/min |
| dprotein | Protein degradation rate | 0.035 | 1/min | [5] | There is no active degradation pathway and that dilution is the dominant way by which it degrades. Rate = ln2/doubling time, where doubling time of strain MG1655 = 20min. Assuming steady-state growth in LB broth as presented in paper. rate = ln2/20 = 0.035/min |
| k2 | Protein production rate (PudA) | 2.2 | 1/min | Please see derivation 2 below. | Please see derivation 2 below. |
| [Arabinose] | Concentration of arabinose | Initial: 0.008 | mM | - | - |
| [mRNA] | Concentration of mRNA | - | mM | - | - |
| [PudA] | Concentration of PudA | - | mM | - | - |
1.Derivation of the maximal expression rate,β
- Average molecular weight (Mw) of a base pair = 660g/mol[6][7]
- Average mass of a base pair = 660g/mol x 1.66x10-24 = 1.1x10-21g
- Volume of an E.coli cell = 1µm3[8] = 1x10-15L
- ∴Mass concentration =
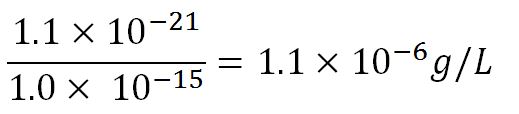
- ∴Molar concentration of 1 base pair in the volume of E.coli =
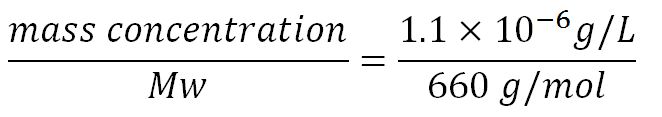 = 1.66x10-6 mM
= 1.66x10-6 mM
- ∴Mass concentration =
- BioBrick assembly plasmid pSB1C3 is a high copy number plasmid (100-300 copies per cell)[9]
- assume 200 copies per cell
- ∴ concentration of the gene per cell = N x 200 x 1.66x10-6mM, where N = number of base pairs
- ∴ concentration of the gene PudA (N = 1644) in the volume of an E.coli cell is = 0.55mM
- Transcription rate in E.coli = 80bp/s[10] = 80 x 1.66x10-6mM/s = 80 x 1.66x10-6 x 60mM/min = 7.97x10-3mM/min
- ∴ Rate of mRNA_pudA production under the control of pBAD = 7.97x10-3 ÷ 0.25 = 0.015/min

- Average molecular weight(Mw) of an amino acid(aa)= 110g/mol[11][12]
- Average mass of an amino acid = 110g/mol x 1.66x10-24=1.83x10-22g/L
- ∴Mass concentration of one aa in the volume of an E.coli =
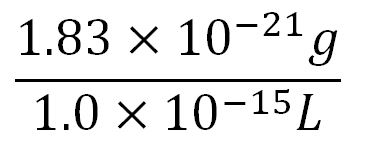 = 1.83x10-6g/L
= 1.83x10-6g/L
- ∴Molar concentration of one aa =
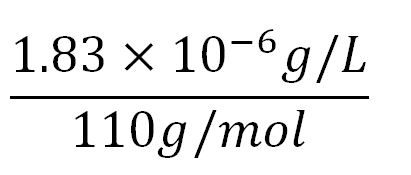 = 1.66x10-5mM
= 1.66x10-5mM
- ∴Mass concentration of one aa in the volume of an E.coli =
- Translation rate = 20aa/s = (20 x 1.66x10-5 x 60)mM/min = 0.020mM/min
- PudA comprises of 548aa[13]
- ∴concentration of PudA's aa in the volume of an E.coli = 1.66x10-5mM x 548 = 9.10x10-3mM
- ∴ Rate of protein production = 0.020 ÷ 4.25x10-3 = 2.2/min


PUR degradation module
The reaction equation of the PUR degradation is:
[Polyurethane]+[PudA]= 4 [ethylene glycol] + 4 [polyisocyanate] + [PudA]
Assumptions:
We assumed 1 mole of polyurethane dispersion can produce 4 moles of ethylene glycol.
The molecular weight of a single polyurethane monomer is 470 g/mol whereas the molecular weight of the polyurethane dispersion is around 2000 g/mol.[14]Therefore, the short-chain polyurethane consists approximately 4 monomers. 4 molecules of ethylene glycol will be produced by degrading one chain of polymer.
We also assumed a simple Michaelis-Menten mechanism for PudA

"[PudA]"is the concentration of the PudA enyme whereas "[Polyurethane]" is the concentration of the polyurethane. The kinetic parameters are defined as below:
| Parameter | Description | Value | Units | Sources | Species | |
|---|---|---|---|---|---|---|
| Km | Michaelis constant | 51.5 | mM | [15] | Comamonasacidovorans | |
| Kcat | Turnover number | 141.75 | 1/min | [16] | Comamonasacidovorans |
Assumptions:
1. The kinetic parameters of PudA are not from the strain E.coli K12 MG1665 we are using. So the assumption here is that the kinetic property of pudA in E.coli is close to that in Comamonasacidovorans
2. Considerations of temperature change and PH change are not included in the model. Therefore, the assumption is that they remain constant during the degradation
Secretion module
The efficiency of Secretion is assumed to be 90% secretion over 2 hours.[17] The rate of secretion in the model is therefore:
rate of secretion = 0.9[concentration of PudA]/120 (mM/mins)
Simulation Results and Industrial Implementation
The simulation result we worked out is especially related to the industrial degradation SRF which we aim to achieve a high degradation efficiency. According to the data we estimated for the percentage Polyurethane in SRF, we have about 5% Polyurethane contained in the waste. Therefore, in the model, we assumed 50g of polyurethane in 1kg of SRF based on the estimation. If we put 1kg of waste into a 5L bioreactor, the approximate concentration of polyurethane would be 5mM. Here is the simulation result:

The modelling result shows that we can efficiently degrade Polyurethane in SRF even in industrial scale. 5mM of polyurethane dispersion can be efficiently degraded in 15 mins. The final concentration of ethylene glycol we produced would be 20 mM that is under the toxicity limitation, this proves that our bio-reactor can maintain the efficiency without the toxicity effects.
As for the implementation of MAPLE system, the polyurethane degradation enzymes in our system are very efficient. Therefore, we need an efficient filter which removing ethylene glycol from the system.
PUR degradation model:Download

2.0 P(3HB) synthesis model ▼
This section is about modelling the plastic-synthesising E.coli developed in our project. The plastic that this module synthesises is the bioplastic poly-3-hydroxybutyrate, also known as P(3HB). The content below shows the process of building, designing and optimising the model. Also, the considerations, assumptions and limitations will be discussed.
The model was built and simulated in Simbiology, a Matlab package designed for modelling biological systems. In our model we have a compartment (labelled "E.coli_1") that represents our engineered cell. The compartment contains the reactions and species that interact with the polymer synthesis pathway.
Here is our model for P(3HB) synthesis. It is part of module 1, this model uses glucose from waste as the input. Here we have the phaCAB operon, which convertsthe metabolite acetyl-coA into poly-3-hydroxybutyrate. We integrated our synthetic pathway into the metabolic pathway. This allowed us to quantitatively predict the P(3HB) production over time. More details about the metabolic model can be found in Module 2.

Firstly, consider the model concerning the synthetic pathway. This model does not take into account the existence of the metabolic pathway present within the bacteria. The synthetic pathway is mainly used for a sensitivity analysis by assuming minimum interference to the cell metabolism. The pathway is rearranged for clarity.
The pathways on the right half of the cell are genetic expressions of the 3 enzymes involved in the plastic synthesis pathway. The pathways on the left represent the actual P(3HB) production pathway.


As can be seen in the Key (above), the yellow, solid circle represents a reaction object and for each object parameter the following needs to be specified for the simulation to work:
- Rate equation or kinetic law (e.g. mass action, Michaelis-Menten etc.)
- Parameters in the rate equation
- Species involved in the reaction
- Value and units for each parameter
Simbiology will then use an ODE solver (ode15 or sundial) to solve these ODEs and give a plot of the specified output(s) (concentration level of a species over time, for instance).
- Note that 3HB deg, Acetoacetate deg, AcetoacetylCoA deg and 3HB-CoA deg are degradation rates (all = 0.035/min, please see assumptions below) of the corresponding species formed along the P(3HB) synthesis pathway.
2.1 Ordinary Differential Equations ▼
Genetic regulations and expressions
BDH2 (3-hydroxybutyrate dehydrogenase)


PhaB (Acetoacetyl-CoA reductase)and PhaC (P(3HB) synthase)


Values, sources and assumptions ▼
| Parameter | Description | Value | Units | Sources | Assumptions/Notes |
|---|---|---|---|---|---|
| β | maximum rate of transcription | 0.032 | mM/min | Please see derivation 1 below. | Please see derivation 1 below. |
| n | Hill coefficient | 2.0 | dimensionless | [18] | For pBAD strong. Taken from the parts registry page. Rounded to 2.0 from 2.26 as Simbiology wouldn't allow a non-integer value for such parameter. |
| K | Activation coefficient | 0.0031 | mM | [19] | For pBAD strong. Taking the "switch point" (from the corresponding parts registry page) as the activation coefficient. |
| dmRNA | mRNA degradation rate | 0.035 | 1/min | [20] | There is no active degradation pathway and that dilution is the dominant way by which it degrades. Rate = ln2/doubling time, where doubling time of strain MG1655 = 20min. Assuming steady-state growth in LB broth as presented in paper. rate = ln2/20 = 0.035/min |
| dprotein | Protein degradation rate | 0.035 | 1/min | [21] | There is no active degradation pathway and that dilution is the dominant way by which it degrades. Rate = ln2/doubling time, where doubling time of strain MG1655 = 20min. Assuming steady-state growth in LB broth as presented in paper. rate = ln2/20 = 0.035/min |
| k2 | Protein production rate (BDH2) | 4.7 | 1/min | Please see derivation 2 below. | Please see derivation 2 below. |
| k3 | Protein production rate (PhaCB) | 0.58 | mM/min | Please see derivation 3 below. | Please see derivation 3 below. |
| [Arabinose] | Concentration of arabinose | Initial: 0.008 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [mRNA] | Concentration of mRNA | - | mM | - | - |
| [BDH2] | Concentration of BDH2 | - | mM | - | - |
| [PhaB] | Concentration of PhaB | - | mM | - | - |
| [PhaC] | Concentration of PhaC | - | mM | - | - |
Derivations ▼
- Average molecular weight (Mw) of a base pair = 660g/mol[22][23]
- Average mass of a base pair = 660g/mol x 1.66x10-24 = 1.1x10-21g
- Volume of an E.coli cell = 1µm3[24] = 1x10-15L
- ∴Mass concentration =
- ∴Molar concentration of 1 base pair in the volume of E.coli = = 1.66x10-6 mM
- ∴Mass concentration =
- BioBrick assembly plasmid pSB1C3 is a high copy number plasmid (100-300 copies per cell)[25]
- assume 200 copies per cell
- ∴ concentration of the gene per cell = N x 200 x 1.66x10-6mM, where N = number of base pairs
- ∴ concentration of the gene BDH2 (N = 768) in the volume of an E.coli cell is = 0.25mM
- Transcription rate in E.coli= 80bp/s[26] = 80 x 1.66x10-6mM/s = 80 x 1.66x10-6 x 60mM/min = 7.97x10-3mM/min
- ∴ Rate of mRNA_BDH2 production under the control of pBAD = 7.97x10-3 ÷ 0.25 = 0.032/min
2.Protein production rate of BDH2, k2
- Average molecular weight(Mw) of an amino acid(aa)= 110g/mol[27][28]
- Average mass of an amino acid = 110g/mol x 1.66x10-24=1.83x10-22g/L
- ∴Mass concentration of one aa in the volume of an E.coli = = 1.83x10-6g/L
- ∴Molar concentration of one aa = = 1.66x10-5mM
- ∴Mass concentration of one aa in the volume of an E.coli =
- Translation rate = 20aa/s = (20 x 1.66x10-5 x 60)mM/min = 0.020mM/min
- BDH2 comprises of 256aa[29]
- ∴concentration of BDH2's aa in the volume of an E.coli= 1.66x10-5mM x 256 = 4.25x10-3mM
- ∴ Rate of protein production = 0.020 ÷ 4.25x10-3 = 4.7/min
3.Protein production rate for PhaB and PhaC, k3
- Relative promoter strengths: J23104 = 1.3RPU, J23101 = 1.0RPU.[30]
- ∴ 104 is 1.3x stronger than 101.
- In absolute units: take GFP synthesis rate (molecules per min per cell) and approximate that as a generic protein synthesis rate for the promoter.
- GFP synthesis rate of 101 = 2232 molecules per min per cell.[31]
- ∴ GFP synthesis rate of 104 = 1.3 x 2232 = 2902 molecules per min per cell
- Assume 1 molecule in an E.coli cell gives a concentration of 1nM.
- ∴ GFP synthesis rate of 104 = 2902 x 1nM = 2.9x10-6nM/min per cell
- Plasmid copy number assumed as 200 (as in derivation 1)
- ∴ GFP synthesis rate of 104 in our E.coli = 200 x 2.9x10-6 = 0.00058nM/min = 0.58mM/min
2.2 Enzyme kinetics ▼
- Note:
- v = velocity of reaction
- Ac-CoA = acetyl-CoA
- AcAc-CoA = Acetoacetyl-CoA
- AcAc = acetoacetate
BDH2 (3-hydroxybutyrate dehydrogenase)
Reaction R1: 
Reaction R1r: 
Values, sources and assumptions
| Parameter | Description | Value | Units | Source | Assumptions/Notes |
|---|---|---|---|---|---|
| kcat,BDH2_f | Turnover number of BDH2 in forward reaction | 22200 | 1/min | [32] | Sequences derived from Pseudomonas fragi but kinetic values from expression and purification of enzymes in E. coli XL1 Blue |
| kcat,BDH2_r | Turnover number of BDH2 in reverse reaction | 7200 | 1/min | [33] | Sequences derived from Pseudomonas fragi but kinetic values from expression and purification of enzymes in E. coli XL1 Blue |
| Ki,NAD+ | Inhibition constant of BDH2 with NAD+ | 2.5 | mM | [34] | Organism Tetrahymena pyriformis |
| Ki,NADH | Inhibition constant of BDH2 with NADH | 1.1 | mM | [35] | Organism Tetrahymena pyriformis |
| Km,AcAc | Michaelis constant for AcAc | 0.37 | mM | [36] | Values in paper reference |
| Km,NAD+ | Michaelis constant for NAD+ | 0.24 | mM | [37] | Values in paper reference |
| Km,NADH | Michaelis constant for NADH | 0.010 | mM | [38] | Values in paper reference |
| Km,3HB | Michaelis constant for 3HB | 0.80 | mM | [39] | source organism Pseudomonas fragi |
| [BDH2] | Concentration of BDH2 | - | mM | - | - |
| [NAD+] | Concentration of NAD+ | initial: 1.6x10-13 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [3HB] | Intracellular concentration of 3HB | 6.4x10-14 | mM | - | External concentration of 3HB assumed to be 0.01g. This is shared by 1.5x1012 cells. Therefore, one cell has 0.01/1.5x1012 = 6.7x10-15 g. To convert it into mol/L: mass of 3HB/molar mass of 3HB = 6.7x10-15/104.1 = 6.4x10-17 M = 6.4x10-14 mM |
| [AcAc] | Concentration of AcAc | initial: 1.0x10-13 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [NADH] | Concentration of NADH | initial: 2.5x10-14 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
atoAD (Acetyl-CoA:acetoacetyl-CoA transferase (α and β subunits))
Reaction R2: 
Reaction R2r: 
Values, sources and assumptions
| Parameter | Description | Value | Units | Sources | Assumptions/Notes |
|---|---|---|---|---|---|
| Vmax,atoAD_f | Maximum rate of atoAD in forward reaction | 0.00244 | mM/min | [40] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| Vmax,atoAD_r | Maximum rate of atoAD in reverse reaction | 0.0108 | mM/min | [41] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| Km,AcAc | Michaelis constant for AcAc | 1.86 | mM | [42] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| Km,Ac-CoA | Michaelis constant for Ac-CoA | 0.26 | mM | [43] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| Km,Acetate | Michaelis constant for Acetate | 53.1 | mM | [44] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| Km,AcAc-CoA | Michaelis constant for AcAc-CoA | 0.035 | mM | [45] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| [AcAc] | Concentration of AcAc | initial: 1.0x10-13 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [Acetate] | Concentration of Acetate | initial: 1.0x10-13 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [AcAc-CoA] | Concentration of AcAc-CoA | initial: 1.0x10-13 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [Ac-CoA] | Concentration of Ac-CoA | initial: 1.0x10-14 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
phaB (Acetoacetyl-CoA reductase)
Reaction R3: 
Reaction R3r:
Values, sources and assumptions
| Parameter | Description | Value | Units | Sources | Assumptions/Notes |
|---|---|---|---|---|---|
| kcat,PhaB_f | Turnover number of PhaB in forward reaction | 6120 | 1/min | [46] | Gene in Ralstonia eutropha was engineered to put in E.coli and then purified |
| kcat,PhaB_r | Turnover number of PhaB in reverse reaction | 3600 | 1/min | [47] | originally in Zoogloea ramigera, expressed in E.coli and then purified |
| Km,NADPH | Michaelis constant for NADPH | 0.15 | mM | [48] | Gene in Ralstonia eutropha was engineered to put in E.coli and then purified |
| Km,AcAc-CoA | Michaelis constant for AcAc-CoA | 0.0057 | mM | [49] | Gene in Ralstonia eutropha was engineered to put in E.coli and then purified |
| Km,NADP+ | Michaelis constant for NADP+ | 0.0060 | mM | [50] | purified from Methylobacterium extorquens |
| Km,3HB-CoA | Michaelis constant for 3HB-CoA | 0.026 | mM | [51] | originally in Zoogloea ramigera, expressed in E.coli and then purified |
| [PhaB] | Concentration of PhaB | - | mM | - | - |
| [AcAc-CoA] | Concentration of AcAc-CoA | initial: 1.0x10-13 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [NADPH] | Concentration of NADPH | initial:3.8x10-14 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [NADP+] | Concentration of NADP+ | initial:1.5x10-13 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [3HB-CoA] | Concentration of 3HB-CoA | - | mM | - | - |
phaC (P(3HB) synthase)
Reaction R4: 
Values, sources and assumptions
| Parameter | Description | Value | Units | Sources | Assumptions/Notes |
|---|---|---|---|---|---|
| kcat,PhaC | Turnover number of PhaC | 1680 | 1/min | [52] | - |
| Km,PhaC | Michaelis constant of PhaC with 3HB-CoA | 0.14 | mM | [53] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| [PhaC] | Concentration of PhaC | - | mM | - | - |
| [3HB-CoA] | Concentration of 3HB-CoA | - | mM | - | - |
atoB (acetyl-CoA acetyltransferase)
Reaction R5: 
Reaction R5r:
Values, sources and assumptions
| Parameter | Description | Value | Units | Sources | Assumptions/Notes |
|---|---|---|---|---|---|
| Vmax,atoB_f | Maximum rate of atoB in forward reaction | 3.8x10-5 | mM/min | [54] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| Vmax,atoB_r | Maximum rate of atoB in reverse reaction | 8.5x10-4 | mM/min | [55] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| Km,atoB | Michaelis constant of atoB with Ac-CoA | 0.47 | mM | [56] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| Km,AcAc-CoA | Michaelis constant for AcAc-CoA | 0.1 | mM | [57] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| Km,CoA | Michaelis constant for CoA | 0.25 | mM | [58] | Enzyme is naturally expressed in E.coli: assume the enzyme is stable during the time course of the simulation. |
| [Ac-CoA] | Concentration of Ac-CoA | initial: 1.0x10-13 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [AcAc-CoA] | Concentration of AcAc-CoA | initial: 1.0x10-13 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
| [CoA] | Concentration of CoA | initial: 5.5x10-13 | mM | see section 2.5 "Initial concentrations of metabolites" | see section 2.5 "Initial concentrations of metabolites" |
Reaction kinetics and mechanisms ▼
2.3 Simulation results ▼
Using the values presented in the tables above, the deterministic model was simulated to see how much P(3HB) could be formed inside the cell. However, with the single cell model, it would be difficult to reflect how much of the plastic could actually be formed on a macroscopic scale. Therefore, modifications to the simulation algorithm were needed to give scaled-up simulation results.
In the scaled-up regime, though, new limitations were encountered. For example, growth, cell division and the concentration gradient that each cell encounters and how much can be taken up by the cells. Given the data available, what could be done was to assume that the bacteria were in a bioreactor, with constant nutrient and oxygen supply, and that they were in the phase of stationary growth (please see assumptions and explanations under the graphs below).
Here, three graphs are presented to show P(3HB)formation over time. The first two graphs are (i)single-cell model results, concentration in mM and (ii) single-cell model results, concentration in g/L. The first graph was the original graph produced from Simbiology as all concentrations were expressed in mM in the model. However, to gain a more intuitive sense of how much could be produced g/L was used. The third graph is the scaled-up simulation result.
(i) please see assumptions and explanations below

(ii) please see assumptions and explanations below

(iii) please see assumptions and explanations below

Graphs (i) and (ii): assumptions and explanations
- To convert from mM to g/L:
- Molecular weight(Mw) of P(3HB) = 1.8x1012g/mol[64]
(Please note that this value was taken from a paper about the "Production of poly(3-hydroxybutyrate-co-4-hydroxybutyrate) in recombinant Escherichia coli grown on glucose" instead of the production of poly(3-hydroxybutyrate). However, this is the closest we managed to get as far as species, substrate and type of plastic are concerned.
Also, it was noticed that the molecular weight would change depending on the cultivation time (as conditions such as growth and pH can also affect it) and it was hard to find a paper that contains the exact same conditions and the range of cultivation times for our simulations. Furthermore, in reality there wouldn't be a single value of molecular weight obtained, but a range of values (known as polydispersity).[65] Therefore, it was assumed that the molecular weight would remain constant during the time course of the simulation and that the value should be considered as an average.)
- ∴P(3HB)concentration in mM x 10-3 x 1.8x1012g/mol = concentration in g/L
Graph (iii): assumptions and explanations
- Assume 1.5x1012cells in a 1L broth, calculated from value obtained from BioNumbers. A range was given: 1-2x109 cells/ml, so 1.5x109 was taken. [66]
- Conditions and considerations accompanying this value are:
- growth medium: LB broth
- Cultivation temperature: 37°C
- wild-type E. coli K-12 strain MG1655 (NB: though not exactly our engineered one, it is the correct strain)
- Conditions and considerations accompanying this value are:
- Assume stationary growth phase.
- Assume that the amount of P(3HB) accumulated during the time course of simulation doesn't exceed the cell's capacity or ability to contain it.
- Same metabolic considerations as above (single-cell results)
- Simbiology single-cell simulation results multiplied by the number of cells stated above to give the red curve.
2.4 Model-guided design and optimisation ▼
Sensitivity analysis: species concentrations
What is sensitivity analysis and why do it?
In order to look at how we could increase the production of P(3HB) we decided to run a sensitivity analysis (also in Simbiology) to identify what species in the model P(3HB) is sensitive to, given the specific conditions with which we set the model. In other words, we calculated the time-dependent sensitivity of P(3HB) with respect to the initial conditions of other species (species formed along the synthesis pathway and enzymes involved).[67]
For a species x, whose concentration depends on time, it can be expressed as x(t). To calculate the sensitivity of x(t) with respect to another species y(t), the sensitivity with normalisation relative to the numerator x(t) can be calculated as:

Therefore, the underlying algorithm for this sensitivity analysis of P(3HB) is:


Sensitivity analysis: enzyme concentrations
The sensitivity analysis of the enzymes involved in the synthetic pathway is carried out, which determine the most sensitive enzyme in the pathway
We plot a time integral of the sensitivity analysis which has the algorithm as:

The senstivity result is:

According to the simulation result, we found that the PHB production is highly sensitive to the concentration of PhaB and PhaC. In contrast, the system is not sensitive to the concentration of atoAD and atoB. In theory, increase the concentration of any of the enzyme will increase the flux of the system. However, for the strain E.coli K12 MG1655, both atoAD (acetoacetate:acetoacetyl-coA transferase) and atoB (acetoacetyl-coA thiolase) favour the reverse reactions, it becomes the rate limiting step in our pathway. Acetoacetyl-coA intends to be converted to acetoacetate instead of 3HB-coA. [68]Therefore, the concentration of PhaB become really critical which push the system to flow in the forward direction.
Scan with different levels of PhaB
The plot below is to show that increasing PhaB concentration within the engineered organism would have a positive effect on the concentration of the P(3HB) synthesised. As so many parameters within the model were taken from different sources, it would be difficult to give a simulation plot that is accurate quantitatively. It was decided to show the single cell simulation for this because the scans were carried out up to the doubling time of 20mins, hence avoided the issue of scaling-up which would have otherwise subjected the simulation to more errors. Therefore, this plot should be interpreted from a qualitative perspective and the trend should be observed.

Difference between promoter expressions after optimisation
In order to choose the strongest promoter that is available to increase the gene expression. We ran a parameter scan of enzyme expression rates of a range of Anderson's constitutive promoters. The simulations shows the PHB production under different Anderson's promoters. The promoters we tested are:
| promoter | biobrick | GFP synthesis rate (mM/min) |
|---|---|---|
| J23100 | [69] | 0.41 |
| J23101 | [70] | 0.45 |
| J23104 | [71] | 0.58 |
| J23118 | [72] | 0.31 |
The derivations of the GFP synthesis rate are:
*Relative promoter strengths: J23104 = 1.3RPU, J23101 = 1.0RPU, J23100 = 0.92RPU, J23118 = 0.76RPU[73]
- In absolute units: take GFP synthesis rate (molecules per min per cell) and approximate that as a generic protein synthesis rate for the promoter.
- GFP synthesis rate of 101 = 2232 molecules per min per cell.[74]
- Assume 1 molecule in an E.coli cell gives a concentration of 1nM.
- ∴ GFP synthesis rate of 101 = 2232 x 1nM = 2.2x10-6nM/min per cell
- Plasmid copy number assumed as 200 (as in derivation 1)
- ∴ GFP synthesis rate of 101 in our E.coli = 200 x 2.2x10-6 = 0.00045nM/min = 0.45mM/min
- ∴ The GFP synthesis rates of other promoters can be calculated by multiplying the RPU value with the GFP synthesis rate of J23101 promoter.
The simulation result:

Although there is just a small increase the gene expression rate, J23104 promoter achieved a much higher PHB production than other constitutive promoters. Therefore, we decided to use J23104 promoter for our improved biobrick BBa_K1149051.
Here is our experimental results that show we have 11 times more PHB after the change of promoter.
 |
P(3HB) synthesis model:Download
Additional script:Download
2.5 Metabolic considerations ▼
In Module 1: Resourceful Plastic, glucose from the degradation is used as the input for the system that produces P(3HB). The P(3HB) synthetic pathway hence needs to be adapted for this purpose. To do so, 3HB monomer would no longer be the source input to the engineered system. So, BDH2 should be removed from the pathway. Acetyl-coA would then become the key resource to produce P(3HB). Also, a metabolic model was needed to show the production of Acetyl-coA from glucose. Furthermore, our synthetic pathway involves several key metabolites from the pathway such as NADPH, NADP+, NADH and NAD+. The initial concentration of those metabolites can be determined from the metabolic model by assuming they are in their steady states. More features and results of the metabolic model can be found in Module 2.
Initial concentrations of metabolites
The model presented in Section 2.0 contains a synthesis pathway that interact metabolic species involved in so many other metabolic reactions elsewhere in the cell. It was, therefore, difficult to determine how much of these metabolites would be left available for our plastic synthesis pathway to utilise. So, a simple metabolic model of the cell (more detail in next paragraph) was needed to simulate and show insight into this problem. We decided to use the steady state levels of these metabolites (without the engineered pathway) and plug the values into the P(3HB) synthesis model in Section 2.0 as the initial concentrations.
The metabolic model involves two pathways - glycolysis pathway and the TCA cycle, where glucose is used as the sole carbon source. The reason is that glucose is directly taken by the glycolysis pathway and there are interactions between Acetyl-coA and TCA cycle. This Simbiology model is based on the one built by Angela Dixon in Predictive Mathematical Model for Polyhydroxybutyrate Synthesis in Escherichia coli.[75]. All reactions and pathways are based on ODEs and kinetics data presented in the original model. It can dynamically predict the concentration of all metabolites involved over a defined simulation time course.
The metabolic model for determining steady state concentrations is shown as below:

Keys:

The initial concentrations of all metabolites and kinetic data are referenced in the paper.[76] The simulation of the metabolites is:

Table of initial and steady-state concentrations of the metabolites:
| Substrates | Initial concentration | Steady state concentration | Units | Sources |
|---|---|---|---|---|
| NADH | 200 | 250 | uM | [77] |
| NAD+ | 1200 | 1600 | uM | [78] |
| Acetyl-coA | 1000 | 100 | uM | [79] |
| Coenzyme A | 250 | 5500 | uM | [80] |
| NADPH | 200 | 380 | uM | [81] |
| NADP+ | 1200 | 1100 | uM | [82] |
<p align="justify"> It should also be noted that acetate, acetoacetate and acetoacetyl-CoA cannot be predicted by the metabolic model. Therefore, their initial concentrations are rough estimates (value similar to other metabolites) such that a reasonable P(3HB) output can be produced.
Assembling the optimised bioplastic producing operon
We made two new improved constructs by modifying the native operon:
The modellers in the drylab predicted that an increased level of pha B enzyme will increase the amount of PHB produced and therefore we decided to change the native Ralstonia eutropha promoter and RBS to a strong constitutive E.coli promoter (J23104) and RBS (0034) in front of the operon. We have succeeded with the swap and built the new biobrick BBa_K1149052 which we refer to as constitutive phaCAB. In parallel, we have also assembled a different configuration of transcription initiation region: the "hybrid". Surprisingly, our results show that our "hybrid" phaCAB construct BBa_K1149051 significantly increased bioplastic production compared to native pha CAB. Please read on to discover more. </p>
We tested the promoter and RBS by cloning them in front of amilCP blue chromoprotein gene (BBa_K1149020). As you can see from the plate below, this expresses a bright blue protein, which became an intrinsic part of our BioNouveau palette.

In order to change the promoter, we designed a forward primer that binds to the beginning of the first gene in the operon and contains a Xba restriction site: Pha_Fw (cgcttctagagatggctactgggaaaggagccg). We used pha_Fw and the suffix primer G1005 primer pain to amplify the insert out of the backbone. The new promoter and RBS were J23104 and B0034 from the biobrick BBa_K608002 We digested the pSB1C3 backbone with this biobrick with SpeI and PstI.
Summary diagram of construction steps:

We performed DpnI digest on the gel purified PCR product (insert) in order to get rid of any contamination from the original plasmid. We did Alkaline Phosphatase treatment of the backbone to remove terminal phosphates in order to decrease self-ligation of plasmid. We transformed the ligation into NEB5 cells and we grew overnight cultures from single cell colonies. The next day, we purified the plasmids and digested 200 ng of them with Xba and PstI to screen for the right clones:

Colony #8 had the expected 4.2kB insert and therefore we sent this plasmid for sequencing. After having the sequenced it, we submitted the new part to registry as BBa_K1149051. This construct has double P+RBS since the native P+RBS stayed there in between the new P+RBS and the operon. We have identified a clone (from colony #6) that contains the new constitutive promoter only and submitted it as BBa_K1149052 biobrick. We are testing both the hybrid and normal constitutive constructs for PHB bioplastic production.
Partial sequence of the constitutive phaCAB construct (BBa_K1149052):

Partial sequence of the hybrid phaCAB construct (BBa_K1149051):

Our chassis (MG1655) survives in mixed waste ▼
These assays were designed to test whether our chassis, E.coli (MG1655) could grow directly with waste over a long period of time.
Waste media
 |  |
Conclusion: MG1655 E.coli are viable and grow on mixed waste alone. Therefore we have established that our chassis could survive in a mixed waste bio-reactor context, which is validation of our concept to industrially implement our system.
Waste conditioned media
These assays were designed to test whether our chassis, E.coli MG1655 strain could grow with waste conditioned media (WCM) over a period of 24-48 hours. Waste conditioned media is a filter sterilised version of the waste media and was designed for several reasons; Firstly we were unsure whether mixed waste would be toxic to E.coli and hence a less concentrated version may be more suitable and secondly large chunks of waste would prevent accurate OD600 measurements and therefore we decided to filter out the largest chunks.
 | |
 |
 |
Conclusion: MG1655 transformed with either empty vector (EV) control or mCherry stress biosensor (BBa_K639003) vector are viable and can grow in waste conditioned media. Therefore waste conditioned media is an appropriate and novel experimental media with which to characterise biobricks within a mixed waste/landfill context. These data are also characterisation of an existing biobrick (BBa_K639003)
Mixed waste degradation: Polyurethane (PUR) ▼
PUR Esterase enzyme activity
Cell lysate assay
Since the PUR Esterases were not secreted, initially the cells were lysed to obtain crude cell extracts in order to test whether the enzymes are active. The Western Blot results showed that the constructs EstCS2 BBa_K1149002, PueB BBa_K1149004 and PulA BBa_K1149006 were being expressed. The cultures expressing these three constructs were grown, lysed by sonication and utilised in a colourimetric assay with the substrate analog para-Nitrophenyl butyrate. The data shows that PUR Esterase EstCS2 BBa_K1149002 is definitely active.
para-Nitrophenyl butyrate (p-NP) is commonly used to indicate an enzyme’s esterase activity. The enzyme cleaves the ester bond and releases the 4-nitrophenol (4-NP), thus causing a colour change from colourless to yellow and an increased absorbance at wavelength 405 nm.

The assay was run in the Eppendorf BioSpectrometer was used to automatically read the absorbance of the reaction mixture every 30 seconds. The concentration of 4-Nitrophenol produced from the reaction was calculated using the Beer-Lambert Law; the extinction coefficient of 4-NP is 18,000 M-1 cm-1 at 405 nm.
The results below show the concentration of 4-NP produced by the three different PUR esterases and compare them to the Empty Vector and Substrate alone as negative controls.


The above graphs clearly show that PUR Esterase EstCS2 is active and cleaving p-NP. The recorded concentrations of 4-NP, in the presence of this PUR Esterase, are much greater than with the Empty Vector or the Substrate alone. Is PUR Esterase EstCS2 active? Yes!


The enzymes expressed by both PueB and PulA do not appear to have esterase activity for this substrate. There is a tiny increase in 4-NP concentration for PueB, PulA, Empty Vector and Substrate alone. This probably indicates that the substrate is slowly degrading by itself. Other constituents of the cell lysates are likely to be causing a slight increase in p-NP degradation, as PueB, PulA and Empty Vector show higher concentrations of 4-NP than the Substrate alone.
Conclusion: PUR Esterase EstCS2 is active!
Mixed waste degradation: Toxicity ▼
Ethylene glycol

Reduced growth at 30°C likely due to decreased efficiency of MG1655 ethylene glycol break down enzymes. These enzymes (see UC Davis 2012) are endogenously expressed and detoxify Ethylene Glycol.
Bioplastic production ▼
E.coli MG1655 containing the PhaCAB construct Part:BBa_K934001[83] accumulates the bioplastic P3HB inside its cells. P3HB can be visualised through staining with Nile Red, which fluoresces strongly in a hydrophobic environment, such as when it binds to the P3HB. However staining will be much lower in cells where P3HB is not present. To see whether P3HB was being produced we grew E.coli containing the phaCAB operon under the native promoter from Ralstonia eutropha. This produced the results we hoped for as seen in the image below.
The P3HB production model indicated that upregulating atoB would lead to an increase in bioplastic production. We changed the promoter before the phaCAB operon to the strong promoter J23104 and a hybrid promoter we created.
The Nile Red plates also provide a preliminary way in which to investigate the effect of changing the promoter to a stronger version. The darker staining of the streaks of the constitutive and hybrid promoter containing constructs suggests the production of higher levels of P3HB in these strains.
 |
We imaged the cells using the fluorescent microscope to qualitatively understand how much P3HB is produced per cell. As indicated by other authors(1), we saw that P3HB is found in localised granules within the bacteria. Empty vector containing cells show little fluorescence. In those that do, the staining is qualitatively different than in the phaCAB containing strains. Nile Red is also used as a lipid stain and this is indicated by its staining of the membranes of EV cells. In contrast, in phaCAB containing cells, staining is restricted to points within the cells.
Each fluorescent image was created by exciting the stain at 530nm for 100ms. Despite this standard treatment of samples the intensity of fluorescence produced by the cells containing the hybrid promoter is greater than from the constitutive ones. This could indicate a difference in the strength of the promoters which lead to phaCAB expression and therefore the amount of P3HB produced. The images also suggest that not every single E.coli is stained. This may be due to the conditions in which the cells were grown or may be dependent upon the stage in the lifecycle of the E.coli.
 |
We extracted the P3HB using a simple technique which maintains the chain length of the P3HB while releasing it from the cells. The amount of plastic extracted from the hybrid promoter-phaCAB(BBa_K1149051[84]) was much larger compared to the native promoter-phaCAB. All other conditions were kept the same.
 |
Increasing Bioplastic Production ▼
We converted the data into the format with which industry compares P3HB production. These are mass of plastic per dry biomass and mass of plastic per litre of culture media. The strain containing the hybrid promoter had a ten fold higher ratio of plastic to biomass than that produced by the original part BBa_K934001
 |
Comparing the amount of plastic produced by the two constructs is visually striking.
| |
Bioplastic from mixed waste ▼
P(3HB) From Mixed Waste
E. coli (MG1655) transformed with native phaCAB (BBa_K934001) were grown with 600ml waste media (12g mixed waste in 600ml M9M- Minimal Media) overnight (16h) at 37oC with shaking. Prior to centrifugation large mixed waste chunks were filtered out. P(HB) was purified as described in Protocols.
.jpg)
See here a video of the process.
3HB Assay: Confirming production of 3HB
We have adapted a medical kit for 3HB detection for chemically analysing our samples. This provided a simple but accurate mean to measure the 3HB liberated from the polymer after phaZ1 depolymerase enzyme treatment. The monomer 3HB indicated the presence of P(3HB) in the material we purified from cells grown on waste as a sole carbon source. Please see the protocol here and our data and calculations here.
 |

We repeated the previously described experiment with E.coli cells containing all three P(3HB) production operons, grown on waste media. We also also tested the material left after the extraction protocol from cells containing the Empty vector only, grown on waste media. The yellow colour indicates presence of 3HB.

Extras ▼
Growth assays with different experimental media
In additional to standard LB and minimal media, several novel experimental media were developed in order to characterise Biobricks within a mixed waste/landfill setting. These media were characterised through an examination of pH and through an array of growth assays with the project chassis, E.coli MG1655 strain.
 |  |
Conclusion: E.coli strain are viable and grow in all of our experimental medias. We have established a novel media that is optimised for characterisation of biobricks within a mixed waste/landfill context.
Protocols ▼
Waste Conditioned Media (WCM)
We added 1g [http://en.wikipedia.org/wiki/Refuse-derived_fuel SRF] (Solid Recovered Fuel) /50 mL LB and then autoclaved the mixture. Once autoclaved, large waste chunks were removed through filter sterilisation (0.2 µm filters) before being used in growth assays as waste conditioned media (WCM). SRF refers to the refuse from recycling facilities that has no value currently and is incinerated to produce power at a cost to the recycling facility, it is composed of 30% plastics while the rest is cellulosic waste
See here how we make up waste conditioned media and purify plastic from it.
Waste Growth Assay
Overnight (O/N) cultures of MG1655 transformed with (BBa_K639003) were diluted to OD 0.05 in either fresh LB or waste conditioned media and plated into 96 well plates (200 µl/well). OD600 was read at the indicated time points and media only (LB) was taken away as background signal. In addition to this a qualitative assay as performed. 9 mL mCherry bacteria were transferred into 300 mL of autoclaved SRF in LB, which were placed in a shaking incubator at 37°C. After 2 days 200 µL of this solution was plated out on chloramphenicol plates, this was then repeated a week later to gauge whether or not the bacteria were still alive. To show that the MG1655 could grow on waste solely, they were grown in PBS (phosphate buffered saline) and autoclaved waste. In 100 mL PBS + waste, 1 mL MG1655 mCherry E.coli were added. They were then grown in a shaking incubator at 37°C and samples were plated after 2 days and 6 days.
MSDS ▼
Some of the reagents we used were slightly toxic to human. In order to reduce the risks of using toxic reagents, we always wear gloves and labcoats. For some toxic reagents, we performed in fume cupboard. Click on the reagents to see the MSDS.
1. Tributyrin
2. Poly(diethylene glycol adipate).
3. Nile red.
4. Arabinose.
6. Acetonitrile.
7. N-Succinyl-Ala-Ala-Pro-Leu p-nitroanilide.
8. Lipase from porcine pancreas.
Future work
We are changing the pelB secretion tag to OsmY-fusion partner for better secretion
As seen in the Western Blot results, some of the enzymes designed for secretion were not secreted. These proteins may have been stuck in the periplasmic space. Thus we are optimising enzyme secretion by building a new construct with OsmY BBa_K892008 fusion for secretion. OsmY is naturally secreted from E.coli and will carry our PUR esterase enzyme with. Once in the culture medium, we think that the enzyme is going to be functional in the fusion form but we included a TEV site in between the domains that can be cleaved by TEV protease if needed.

The linker sequence we have designed between the domains is T G S E N L Y F Q G S (ACCGGCAGCGAGAACCTGTACTTCCAAGGCAGC) and includes the 7 amino acids of the TEV site as well as 4 other amino acids for flexibility.
This construct will be a substitute for BBa_K1149002 which is the PUR esterase EstCS2 with the pelB seretion tag.
UPDATE 28-10-13:
Whilst further optimisation is required a western blot analysis of our new construct OsmY-ESTC2 indicated that our enzyme maybe secreting into the supernatant.
We will test our Hybrid phaCAB within a bioreactor setup
We will inoculate a 15L bioreactor at Imperial College London to perform a pilot scale test of our plastic producing system.
{kind=link}