 "
"
Team:XMU Software
From 2013.igem.org
YuezhenChen (Talk | contribs) |
YuezhenChen (Talk | contribs) |
||
| Line 10: | Line 10: | ||
<div id="xmu_body"> | <div id="xmu_body"> | ||
<div id="xmu_software"> | <div id="xmu_software"> | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
<!--导航菜单--> | <!--导航菜单--> | ||
<div id="title"> | <div id="title"> | ||
Revision as of 11:35, 14 September 2013
<!DOCTYPE HTML>






XMU-Software-2013 team consists of 11team members, 2 instructors and 3 advisors. This extraordinarily energetic and creative team is tightly connected by trust, collaboration and affection among its members. Learn more about our team on team page.
Our project includes 2 independent software tools-the brick worker and E’NOTE. The former is a software suit for the evaluation and optimization of biobricks, i.e., promoter, RBS, protein coding sequence and terminator. E’NOTE is a web application serveing as an assistant for experiments. It’s useful functions such as experiments recording and experimental template customization make experimental process easier and more enjoyable. To learn more about our project, please visit Project page.
See more about XMU-Software-2013 safety form on safetyform page
XMU-Software-2013 has hosted a series of activities aiming at facilitating communication and collaboration among iGEM teams, propagating iGEM competition as well as promoting the development of synthetic biology. And these efforts are well paid off and have received satisfying feedback. More information is available at Outreach page.
See more about XMU-Software-2013 Notebook on Notebook page.





































































Dr. Bai-shan Fang is the professor in College of Chemistry & Chemical Engineering at Xiamen University. In the group of biocatalysis and biotransformation, his research mainly focuses on synthetic biology, mining and the transformation of the enzyme, construction of bio-molecular machines, the application of new biocatalysts. His major role of XMU iGEM team is to mentor for all and to enlighten the members.

Dr. I-Son Ng is the associate professor in College of Chemistry & Chemical Engineering at Xiamen University. Her research interests are biofuel, engineering of enzyme and protein, zymology, genetic engineering, biochemical separation procedures and proteomics. Her role for the project is to provide suggestions and instruments.

It is great for a team to have an omniscient advisor, even better an inspiring one. Ruosang Qiu , our beloved advisor, definitely is offering both. Her hard work as well as undoubted adorable personality is the motivation of our team members’ efforts, her clear mind combined with provident planning lays the foundation of our successful project, To quote her words: I’ was a happy iGEMer in 2012, I’m going to make you all happy iGEMers in 2013.

The past three years have seen his tremendous dedication to iGEM, Xin Wu, a passionate team member in XMU-China-2011, a devoted team leader in XMU-China-2012 and now, an invaluable advisor in XMU-Software-2013. Had it not been Xin Wu's constant encouragement and guidance, we,the inexperienced iGEMers might have been faint-hearted and failed to face up to the challenges during the process. It is drawing on his expertise in synthetic biology and proficiency in iGEM competition that we have solved the seemingly unsolvable and conquered the seemingly unconquerable.

Youbin Mo is one of the great advisors of XMU software team in 2013. As a computational biophysicist, y, he is an unquestionable master of biological model and computer programing. In the meanwhile, website constructing is also Mo’s technical ability which he acquired by participating the iGEM last year. Youbin gives play to his talent by teaching fundamental program skills to new iGEMers as well as directing them to be self-reliant synthetic biologists.
All work described on this wiki or on our parts registry pages was done by iGEM Team XMU-China 2012. We managed the whole project, from planning to financing the complete lab work by ourselves. Nevertheless we could not have done all this work without the help, advice and guidance of several people. Therefore, special thanks to the following people:
Our advisor Graduate Xuesong Zheng for helping us in fluorescence test and immobilization.
Prof. Xiaodong Chen for providing us many instruments such as ELISA reader to test the fluorescence and his students Song Huang, Renpan Deng, Xiang You for experimental guidance.
Prof. Yinghua Lu and his student Shiduo Zhang for helping us make the PDMDAAC-NaCS microcapsules.
Dr. Ying Zeng, Kunming Institute of Botany, Chinese Academy of Sciences, who has provided us with valuable guidance in constructing gene circuits.
Xiamen, also known as Amoy to the west, is a cozy city located in the southeastern part of China, and has a relaxing coastal charm with a population of 1.3 million. It's a historical harbor city which was founded in the mid-14 century, in the early years of the Ming Dynasty. In the early 1980's, Xiamen was declared as one of China's first Special Economic Zone, taking advantages of the city's heritage as a trading center and the proximity to Taiwan. In 2004 the city won the finals of the world's Human Settlements and Environment Award, "Nations in Bloom". Xiamen is one of China's most attractive and best-maintained resort city, and attracts a large number of foreign and local tourists. The city is easily accessible by air, and there are direct flights from Hong Kong, Kuala Lumpur, Osaka, Seoul, Singapore and Tokyo. Within China, Xiamen airport is linked to more than 30 domestic airports.


Xiamen University (XMU), also known as Universitas Amoiensis in Latin, is one of the top universities in China. It was founded in 1921 by Tan Kah-Kee, the well-known patriotic overseas Chinese leader. As an integrated university, XMU owns a comprehensive branches of discipline as well as many specialized institutes. Economy, counting, chemistry, life science and marine science all win high fame nationwide and even worldwide. The main campus of XMU locates in a picturesque setting between the sea and a scenic mountain, spreading over 150 hectares, and is generally regarded as the most beautiful campus in China.
Gene expression in both prokaryotes and eukaryotes is frequently controlled at the level of transcription. This process can be represented as a cycle consisting of four major steps: (1) promoter binding; (2) RNA chain initiation; (3) RNA chain elongation; and (4) termination. Since regulatory controls are exerted at each step, an understanding of the mechanism of each step is of general importance in understanding gene expression.
In the promoter part of our program, we’ve discussed the mechanism of promoter binding step and how it affects the transcription level. To complete our biobrick evaluation program and to better understanding of transcription process, we integrated the software developed by 2012 iGEM team SUSTC-Shenzhen-B to realize the prediction of transcription termination efficiency.
Termination, the last step of the transcription cycle, occurs when the RNA polymerase releases the RNA transcript and dissociates from the DNA template. It is important that transcription is imperfectly terminated at some terminator so that the ratio of the amount of the mRNA transcribed from upstream and that from downstream of the terminator is controlled. This regulation is quantified by the termination efficiency (%T),
Two mechanisms of transcription termination and two classes of termination signals have been described in bacteria: rho-dependent and rho-independent.
Rho-independent (also known as intrinsic) terminators are sequence motifs found in many prokaryotes that cause the transcription of DNA to RNA to stop. These termination signals typically consist of a short, often GC-rich hairpin followed by a sequence enriched in thymine residues.
The conventional model of transcriptional termination is that the stem loop causes RNA polymerase to pause and transcription of the poly-A tail causes the RNA: DNA duplex to unwind and dissociate from RNA polymerase.
In 2011, iGEM team SUSTC-Shenzhen-B developed a software tool TTEC to predict terminator efficiency. It takes DNA sequences as input and returns the terminator efficiency value.
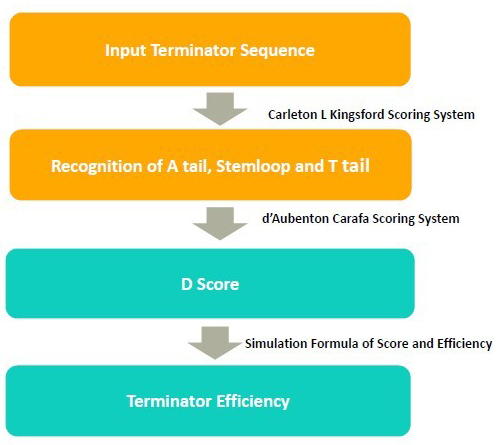
In the algorithm, it takes 3steps to calculate the terminator efficiency:
1. Use RNA folding algorithm to predict the secondary structure of terminator and and recognize A tail, stemloop and T tail.
2. From the secondary structure, we calculate the free energy of stem loop, and generate a score by considering stem loop free energy and T tail
3. From the score, we predict the terminator efficiency based on the score-terminator equation.
The prediction of secondary and recognition of A tail, stemloop and T tail are achieved by Kings ford scoring system.
In 2007, Carleton L Kingsford et al described TransTermHP1, a new computational method to rapidly and accurately detect Rho-independent transcription terminators.
They put forward an algorithm to predict Rho-independent terminators.The first 15 bases of the potential tail sequence are scored using a function:

where

for n=1...15 and =1.
The energy of potential hairpin configurations adjacent to a reference position can be found efficiently with a dynamic programming algorithm. The table entry hairpin_score[i,j] gives the cost of the best hairpin structure for which the base of the 5' stem is at nucleotide position i and the base of the 3' stem is at position j. The entry hairpin_score[i,j] can be computed recursively as follows:

The function energy(i,j) gives the cost of pairing the nucleotide at i with that at j, and loop_pen(n) gives the cost of a hairpin loop of length n. The hairpin's loop is forced to have a length between 3 and 13 nt, inclusive, by setting loop_pen(n) to a large constant for any n outside that range. The constant 'gap' gives the cost of not pairing a base with some base on the opposite stem and thus introducing a gap on one side of the hairpin stem.
Table 1
Parameters used to evaluate hairpins
Pairing Energy
G-C -2.3
A-T -0.9
G-T 1.3
Mismatch 3.5
Gap 6.0
Loop_pen(n) 1•(n - 2)
Parameters used to evaluate the energy of a potential hairpin where n is the length of the hairpin loop
The D score is calculated by Carafa Scoring System
Scoring System 2 is based on the model created by d'Aubenton Carafa2 .The score of terminator consists of two parts, the free energy of stemloop and the score of 15 nt poly T tail. The free energy of stemloop is calculated using Loop Dependent Energy Rules3. The minimization of the free energy also determined the secondary structure of the stemloop. T tail score is calculated by the formula given by d’ Aubenton Carafa.
Detailed Calculation of Score
i. Closing Base Pair
For an RNA sequence, we number it from 5’ to 3’ . If i < j and nucleotides ri and rj form a base pair,we denote it by i.j. We call base ri’ or base pair i’.j’ is accessible from i.j if i <i’ ( <j’ ) <j and if there is no other base pair k.l so that i <k <i’ ( <j’ ) <l <j. We denote the collection of base and base pair accessible from i.j by L(i,j). Then i.j is the closing base pair. Here “L” means loop.
ii. n-loop
If the loop contain n – 1 base pairs, we denote it by n-loop. (Because there is a closing base pair, so we denote it by n-loop even though the closing base pair is not included in the loop.)
Here we can divide loops which may be formed in the terminator secondary structure into two kinds.
1-loop : Hairpin loop(size of loop shouldn’t be smaller than 3)
2-loop : Interior Loop(right strand size and left strand size are both bigger than 0.)
Buldge(Size of one strand is bigger than 0 and that of another strand is 0.)Stack(size of the loop is 0.)
G(i,j)= min { GH ( i , j ) , GS( i , j ) + G ( i + 1 , j – 1 ) , GBI( i , j ) } ;
GBI ( i , j ) = min{ gbi( i , j , k , l ) + G( k , l ) } for all 0 < k – i + l – j - 2 < max_size
G(i,j) is the minimum free energy change of stemloop formation. GH is the free energy change to form a hairpin loop. GS is the free energy change to form a stack. GBI is to calculate the minimum free energy change of structure containing 2-loop. gbi(i,j,k,l) is the free energy change to form 2-loop.
Here we consider 15 nucleotide in the downstream of stemloop. T tail score nT is calculated as follows :

In our program, if the length of the T tail( n ) is less than 15, we will only consider n nucleotides. If TL is more than 15, we will only consider 15 nucleotides.
Score = nT * 18.16 + deltaG / LH * 96.59 – 116.87
Here nT is T tail score. deltaG is the minimum free energy change of stemloop formation. LH is the length of stemloop5.6























 is the target ratio of k-th codon,
is the target ratio of k-th codon,  is the actual ratio of k-th codon in the sequence,the best value of cpi is 0.2 in the software.
is the actual ratio of k-th codon in the sequence,the best value of cpi is 0.2 in the software.


 stands for the ratio of single codon ckin the complete genome'
stands for the ratio of single codon ckin the complete genome' is the number of pair (ci,cj) in high-expression genes,and high-expression genes are genes whose copy numbers of mRNA can be detected at least 20 per cell.
is the number of pair (ci,cj) in high-expression genes,and high-expression genes are genes whose copy numbers of mRNA can be detected at least 20 per cell. 
 equals to the number of amino acid encoded by ci in the whole protein set.
equals to the number of amino acid encoded by ci in the whole protein set. 











 and
and  , the domination status can be evaluated as follows:
, the domination status can be evaluated as follows: >
> and
and  >=
>= , sequence 1 dominates sequence 2. >= and > , sequence 1 dominates sequence 2.< and <= , sequence 2 dominates sequence 1. <= and < , sequence 2 dominates sequence 1.
, sequence 1 dominates sequence 2. >= and > , sequence 1 dominates sequence 2.< and <= , sequence 2 dominates sequence 1. <= and < , sequence 2 dominates sequence 1.
| Part number. | Where did you get the physical DNA for this part (which lab, synthesis company, etc) | What species does this part originally come from? | What is the Risk Group of the species? | What is the function of this part,in its parent species ? | |
| Ex | BBa_C0040 | Synthesized, Blue Heron | Acinetobacter baumannii | 2 | Confers tetracycline resistance |
| 1 | BBa_K1070000 | PCR, Dr.Baishan Fang's lab, Xiamen University | Registry of Standard Biological Parts | 1 | The promoter induced by arabinose |
| 2 | BBa_K1070001 | PCR, Dr.Baishan Fang's lab, Xiamen University | Registry of Standard Biological Parts | 1 | The promoter induced by arabinose |
| 3 | BBa_K1070002 | PCR, Dr.Baishan Fang's lab, Xiamen University | Registry of Standard Biological Parts | 1 | The promoter induced by arabinose |
| 4 | BBa_K1070003 | PCR, Dr.Baishan Fang's lab, Xiamen University | Registry of Standard Biological Parts | 1 | The promoter induced by arabinose |
| Species | Strain no/name | Risk Group | Risk group source link | Disease risk to humans? If so, which disease? | |
| Ex | E.coli(K 12) | NEB 10 Beta | 1 | www.absa.org/riskgroups/bacteria search.php?genus=&species=coli | Yes. May cause irritation to skin, eyes, and respiratory tract, may affect kidneys. |
| 1 | E.coli(K 12) | DH5α | 1 | http://www.absa.org/riskgroups/bacteriasearch.php?genus=Escherichia | Yes. May cause irritation to skin, eyes, and respiratory tract, may affect kidneys. |
| 2 | E.coli(B) | BL21 | 1 | http://www.absa.org/riskgroups/bacteriasearch.php?genus=Escherichia | Yes. May cause irritation to skin, eyes, and respiratory tract, may affect kidneys. |









