 "
"
Team:Grenoble-EMSE-LSU/Documentation/Notebook/June
From 2013.igem.org
(Difference between revisions)
| (41 intermediate revisions not shown) | |||
| Line 18: | Line 18: | ||
<li id="titre"> | <li id="titre"> | ||
<h1>June</h1> | <h1>June</h1> | ||
| - | + | <h2>Week 1 (3-7)</h2> | |
<h3>Monday</h3> | <h3>Monday</h3> | ||
| - | + | ||
| + | <p> | ||
| + | We observed that absorbance measurements differed according to the device used to make them. For example, the Tristar microplate reader and the Thermo Electron Corporation Genesys 6 spectrophotometer we were using gave different OD600 readings for the same samples. This can be explained by the fact that OD600 measurement values come from diffusion rather than actual absorbance, and thus the distance from the sample to the detector is a factor in the reading. Since it is likely these distances are different from one machine to the other, it would explain the differences in measurement.<br><br> | ||
| + | We are then confronted with a dilema: which machine do we trust when referring to literature that mentions OD600 values? We settle that an OD600 reading corresponds to a concentration of bacteria in a culture. We then need to establish the relationship between this concentration and the OD600 reading for both machines. For this we will use cell counting chambers and a microscope to count the cells in a small sample volume. Multiple readings will allow us to eliminate the significant noise that stems from such a measurement. The target is around 100 cells per counting chamber in order to get statistical significance without having to count too many cells.<br><br> | ||
| + | We know that the OD600 curve is linear up to A=0.5 . With values beyond this threshold, the curve levels off and measured values are lower than actual values.<br><br> | ||
| + | </p> | ||
| + | <h3>Tuesday</h3> | ||
<p>Preparation of freezable competent cells (M15): 40 alicos of 100µL</br> | <p>Preparation of freezable competent cells (M15): 40 alicos of 100µL</br> | ||
Test of the competent cells.</p> | Test of the competent cells.</p> | ||
| + | <p>Redaction of a guide for the future grenoble iGEMers, to explain how to manage the association | ||
| + | </p> | ||
| + | <p align="CENTER"> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/1/1c/Grenoble_sarah_travaille.jpg"width="300px" /> | ||
| + | </p><p id="legend">Sarah working on the association guide.</p> | ||
<h3>Wednesday</h3> | <h3>Wednesday</h3> | ||
| - | <p> | + | <p>Beginning of ‘intra-cell ROS concentration’ modelisation.</br> |
| - | + | </br> | |
| + | Preparation of competent cells via <a href="https://static.igem.org/mediawiki/2013/f/fe/Grenoble_Protocols-TSS_Transformations.pdf">the TSS method </a><br> | ||
| + | This method gives cells that are more competent than with CaCl2, but the reagents are harder to prepare. The cells can be stored at -80°C like CaCl2-competent cells.<br><br> | ||
Transfection of competent cells with the biobricks :</br> | Transfection of competent cells with the biobricks :</br> | ||
- pLac</br> | - pLac</br> | ||
| Line 32: | Line 45: | ||
- RBS (Elowitz 1999)</br> | - RBS (Elowitz 1999)</br> | ||
- GFP</br> | - GFP</br> | ||
| - | + | </br> | |
| + | </dl> | ||
First contact with Arduino : the electronic device that will be used as a controler, just as the Cambridge 2010 iGEM team. </br> | First contact with Arduino : the electronic device that will be used as a controler, just as the Cambridge 2010 iGEM team. </br> | ||
| Line 39: | Line 53: | ||
</p> | </p> | ||
<h3>Thursday</h3> | <h3>Thursday</h3> | ||
| + | <p>Failure with the transfections.</br> | ||
| + | Other problem with the petri dishes : everything grew, even the WT colonies that should have died because of antibiotics. Maybe the antibiotics are too old since they were used by Grenoble iGEM Team of 2012.<br><br> | ||
| + | Another note: since M15 cells were used last time and contain the pREP4 plasmid, this plasmid may prevent isolation of other plasmids of similar sizes during electrophoresis after minipreps with transformed M15 cells. It is then useful to make a stock of competent wild-type cells without extra plasmids. Extra care must be taken when preparing these types of competent cells since there is no antibiotic in the solution, and so contaminations are more likely to happen.<br><br> | ||
| + | Used cells were BW25113.<br><br> | ||
| + | Note: cells were vortexed during the preparation to resuspend them but this is apparently not recommended at all! They may have been damaged by this treatment and thus the preparation will have to be made again.</p> | ||
<h3>Friday</h3> | <h3>Friday</h3> | ||
| - | < | + | <p> |
| - | <h2>Week 2</h2> | + | Preparation of freezable competent cells (WT)(40 alicos of 100µL)<br><br> |
| + | The protocol is available on the team wiki.<br><br> | ||
| + | The cells used were M15 cells featuring the pREP4 lacI-producing plasmid (M15 cells are an expression strain used to express recombinant proteins from the lactose promoter. LacI prevents expression until the researcher introduces IPTG into the culture. Since most strains do not produce enough lacI for good repression, this plasmid makes more so that repression is efficient). The pREP4 plasmid confers kanamycin resistance.<br><br> | ||
| + | Same transfections than wednesday, with fresh antibiotics.</br> | ||
| + | </br> | ||
| + | PCRs of pAraBad, mCherry and KillerRed, only pAraBAD worked.</br></br> | ||
| + | <strong>Team meeting</strong></br> | ||
| + | TODO List :</br> | ||
| + | - Find a name</br> | ||
| + | - Begin construction of :</br> | ||
| + | ->pLac-RBS-YF1-fixJ</br> | ||
| + | ->pFixK2-RBS-GFP</br> | ||
| + | - glycerol-stock BBs</br> | ||
| + | - E-glometer, caracterisation of LED</br> | ||
| + | </p> | ||
| + | <h2>Week 2 (10-14)</h2> | ||
<p> | <p> | ||
Little Summary with Keywords | Little Summary with Keywords | ||
| Line 48: | Line 82: | ||
<h3>Monday</h3> | <h3>Monday</h3> | ||
| - | < | + | <p>PCRs of mCherry and KR (did not work). Is there a problem with the primers or with the DNA templates? Need to work on that</br> |
| + | Put the transformed cells on petri dishes with the right antibiotics</br> | ||
| + | Receive the TSL230RD photodiodes</br> | ||
| + | Begin the test of Arduino (digital output) by turning on and off a LED and using a Graphical User Interface (GUI) to control the intensity.</br></br> | ||
| + | <strong>Construction of the pQE30::KillerRed vector</br></strong> | ||
<p> | <p> | ||
| - | Incubated a 50mL overnight culture of E. coli M15[pRep4] cells (Qiagen, Venlo, Netherlands), containing the pQE30::αSNAP vector, in preparation for midiprep. | + | Incubated a 50mL overnight culture of <em>E. coli</em> M15[pRep4] cells (Qiagen, Venlo, Netherlands), containing the pQE30::αSNAP vector, in preparation for midiprep.<br><br> |
| + | Several of our cultures show signs of contamination with different organisms than E. coli. This is very detrimental to cultures as there can be contamination of DNA in minipreps, the cultures don't grow as well if there is competition and it makes physiological studies of the bacteria impossible.<br><br> | ||
| + | One of the contaminated cultures shows antibiotic resistance that it shouldn't have. This is perhaps caused by bad antibiotics that have degraded with bad storage conditions. The antibiotic stocks for ampicillin, a beta-lactamase-type antibiotic that is particularly prone to degradation over time, were replaced. | ||
| + | |||
| + | |||
</p> | </p> | ||
<h3>Tuesday</h3> | <h3>Tuesday</h3> | ||
| - | < | + | <p><strong>Construction of the pQE30::KillerRed vector</br></strong> |
| - | < | + | |
| - | Midi-prepped pQE30::αSNAP. The DNA sample concentration was only of 10 ng/µL. The experiment has to be re performed. < | + | Midi-prepped pQE30::αSNAP. The DNA sample concentration was only of 10 ng/µL. The experiment has to be re performed. </br> </br> |
| - | Incubated a 10mL overnight culture of E. coli M15[pRep4-pQE30::αSNAP] cells, in preparation for miniprep. | + | Incubated a 10mL overnight culture of <em>E. coli<em> M15[pRep4-pQE30::αSNAP] cells, in preparation for miniprep. |
</p> | </p> | ||
| + | <p>- Study the reasons why the PCR of mCherry and KR did not work.</br> | ||
| + | - New failure of the petri dishes, everything grew => miniprep the WT and found that there is already something in it.</br> | ||
| + | - These WT are not really WT, they can’t be used! We may have switched the real tube of WT with another one. Preculture WT from a glycerol stock to do again the transfection.</br> | ||
| + | - Create the first elements of an interface for the electronic part with <I>Processing</I> and <I>Arduino</I>.</p> | ||
<h3>Wednesday</h3> | <h3>Wednesday</h3> | ||
| - | < | + | |
| - | < | + | <p><strong>Construction of the pQE30::KillerRed vector</strong></br> |
Mini-prepped pQE30::αSNAP/pRep4 and got a 98,9 ng/µL DNA sample.<br/> <br/> | Mini-prepped pQE30::αSNAP/pRep4 and got a 98,9 ng/µL DNA sample.<br/> <br/> | ||
| - | Digested pQE30::αSNAP with BamHI and KpnI restriction enzymes. Separation of the gene of non interest (αSNAP) from the pQE30 vector backbone by gel electrophoresis (1.2 % agarose, 30 min, 135V).<br/> <br/> | + | <a href="https://static.igem.org/mediawiki/2013/e/e3/Grenoble_Digestion_ofpQE30alphaSNAP.pdf">Digested pQE30::αSNAP with BamHI and KpnI restriction enzymes.</a> Separation of the gene of non interest (αSNAP) from the pQE30 vector backbone by gel electrophoresis (1.2 % agarose, 30 min, 135V).<br/> <br/> |
</p> | </p> | ||
<p align="CENTER"> | <p align="CENTER"> | ||
<img src="https://static.igem.org/mediawiki/igem.org/b/b0/PQE30alphaSNAP_110613.jpg" alt="restriction of pQE30::alphaSNAP with KpnI and BamHI 11/06/13" width="600px" /> | <img src="https://static.igem.org/mediawiki/igem.org/b/b0/PQE30alphaSNAP_110613.jpg" alt="restriction of pQE30::alphaSNAP with KpnI and BamHI 11/06/13" width="600px" /> | ||
| + | </p> | ||
| + | <p> | ||
| + | </br> | ||
| + | - Add some element on the interface of Processing</br> | ||
| + | - Write on computer a code that turn on one led or another according to the toggle that is pushed on the interface and generate a graph according to the intensity of the light represented by the value of a slider.</br> | ||
</p> | </p> | ||
<h3>Thursday</h3> | <h3>Thursday</h3> | ||
| + | <p>- Weld electric wires on the Surface Mounted Device (SMD) photodiode to test it on the wordkbench</br></p> | ||
| + | |||
| + | <p style="margin:20px" align="CENTER"> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/3/3d/Photodiode_weld.png" alt="SMD_Photodiode_Wire" width="600px" /> | ||
| + | </p> | ||
| + | |||
| + | <p>- Read the datasheet of the photodiode (cf Electronic document)</br> | ||
| + | - Learn how to record the signal of the photodiode with Arduino - thanks to a tutorial on <a href="http://bildr.org/2011/09/tsl230r-arduino/">this website</a></br> | ||
| + | - Add some element on the interface of Processing</br> | ||
| + | - Test the analog input of Arduino</br> | ||
| + | - Verification of all the parameters for the non-working PCRs, list of test to perform</br> | ||
| + | </p> | ||
<h3>Friday</h3> | <h3>Friday</h3> | ||
| - | <h2>Week 3</h2> | + | <p>- PCR with tests (pQE30, and mCherry 2012) on mCherry-pAraBAD using a new sample of mCherry DNA.</br> |
| - | <h2>Week 4</h2> | + | The test worked, its seems like the problem comes from the primers, I will perform cross pcr next week.</br> |
| - | + | - Gel extraction on KillerRed.</br></br> | |
| - | + | <strong>Team Meeting</strong> | |
| - | + | </p> | |
| - | + | <h2>Week 3 (17-21)</h2> | |
| - | + | <h3>Monday</h3> | |
| - | + | <p>- PCR on KillerRed-Pbad</br> | |
| + | - Weld electric wire on a new SMD photodiode to test it on the wordkbench - because the first one didn't appreciate to have 5V on its output instead of its input.</br> | ||
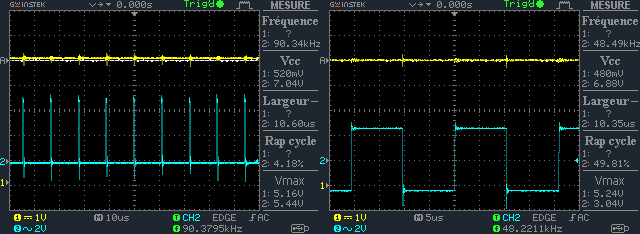
| + | - Test the response of the photodiode for different light powers (irrandiance µw/cm²), sensitivities and frequence scalings (cf <a href="https://static.igem.org/mediawiki/2013/5/51/Oscilloscope.png">oscillogram</a>).</br> | ||
| + | - The 50% duty cycle mode may be easier for the algorithm in Arduino than the pulse train mode because each output pulses last 2*1/foutput.</br> | ||
| + | - Program Arduino and the Processing interface for the photodiode. Test it. The graph on the interface fits the irrandiance given by the oscilloscope</br> | ||
| + | </p> | ||
| + | <h3>Tuesday</h3> | ||
| + | <p>- MIDIPrep with PQE30 plasmids | ||
| + | - PBad-KillerRed overlapping PCR -> failed</br> | ||
| + | - <strong>Weekly reunion decisions</strong> :</br> | ||
| + | - Electronics : wait to know what/how/when to measure / Create a "general and easily changeable" program.</br> | ||
| + | - Webdesign : still in discussion (‘Areva Style’ with improvements ?)</br> | ||
| + | </p> | ||
| + | <h3>Wednesday</h3> | ||
| + | <p>- New PCR on pBAD_KillerRed to have larger amount of DNA for the Overlapping PCR</br> | ||
| + | - Compare the different mode of the photodiode (duration pulse -500ns or duty cycle of 50%)</br> | ||
| + | - Use the oscilloscope to know if the program gives the same results - seems to have some difficulties for high frequencies.</br> | ||
| + | - Creation of models taking account of the inertia of some quantity : measure of fluorescence with severals half life for the GFP</br> | ||
| + | - <strong>2 kind of controls :</strong> </br> | ||
| + | - On/Off control, it is not bad</br> | ||
| + | - PID regulator : it is very bad, don’t achieve to stabilize anything</br> | ||
| + | ->We need to develop a predictive control</br></p> | ||
| + | <h3>Thursday</h3> | ||
| + | <p>- Calculate the standard deviations of the irradiance for different number of periods used for the measurement and the standard deviation for 100 periods for different irradiance.</br> | ||
| + | <strong>Conclude</strong> that 100 periods is the most efficient number of periods, it allows to take a measurement every 5 min and has a low standard deviation - error lower than 0.5%.</br> | ||
| + | </p> | ||
| + | <h3>Friday</h3> | ||
| + | <p><strong>Team meeting</strong></br></br> | ||
| + | - Characterise the complete response of the Arduino’s program thanks to a function generator - from less than 1Hz to more than 500kHz.</br> | ||
| + | - Found that Arduino miss some pulses after 37kHz, after this critical frequency the calculated frequencies is divided by two.</br> | ||
| + | - Cannot calculated frequencies under 1Hz because of the architecture of the program (long = natural number) | ||
| + | </p> | ||
| + | <h2>Week 4 (24-28)</h2> | ||
| + | <h3>Monday</h3> | ||
| + | <p>- Look for information on high power LED and shops that are selling these in Grenoble.</br> | ||
| + | - Look for electronic circuit that can be used to control the intensity of LED -> we find a Dimmer on <a href="http://www.roboticus.org/electronique/52-controler-une-led-de-puissance">this french website</a> that seems to fit our condition. <I>(at the beginning there was only two diodes instead of three and there was a potentiometer instead of the two resistors, these changes will be explained later)</I></p> | ||
| + | <p align="center"><img src="https://static.igem.org/mediawiki/2013/f/f2/Highpowerled.png" alt="Dimmer" width="300px" /> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/f/fe/MR16_COB5W_01.jpg" alt="LED Lamp" width="300px" /> | ||
| + | </p> | ||
| + | <h3>Tuesday</h3> | ||
| + | <p>- Buy high power LED from TopLED, power supply (12V) and accessories</br> | ||
| + | - Test the LED and the power supply, test the Dimmer with the PWM from the function generator. It seems to work well but the potentiometer seems not to be a good choice to lock the intensity.</br> | ||
| + | - Look for an optic system to measure fluorescence or the project. | ||
| + | </p> | ||
| + | <h3>Wednesday</h3> | ||
| + | <p>- Use Arduino and a slider with Processing instead of the function generator - work on the algorithm to control the intensity of the light thanks to Arduino. It seems to work but the components of the electronic circuits need to be changed for more efficient one. | ||
| + | - Transform XL1 Blue and M15 with pQE30 to test the transformation efficiency of the strains. | ||
| + | - Plate 90% on one Petri dishes and 10% on others for each strains | ||
| + | </p> | ||
| + | <h3>Thursday</h3> | ||
| + | <p>- Take picture of the new PCR with the right primers - KR seems to be amplified.</br> | ||
| + | - The transformation with XL1 Blue seems to be more efficient than with M15, there are more colonies for the XL1 Blue than for M15 but we notice that they are smaller. | ||
| + | - Transform XL1 with mCherry and the different ligation products. | ||
| + | - Put strains with luciferase plasmid, pJT122 and pPLPCB(S) in culture, do glycerol stocks - tomorrow we need to miniprep the Voigt’s plasmid and then transform XL1 Blue with the two plasmids to begin the test of the light sensitive promoter.</p> | ||
| + | <h3>Friday</h3> | ||
| + | <p><strong>Team meeting</strong></br></p> | ||
</li> | </li> | ||
| + | |||
| + | <li id="next"><a href="/Team:Grenoble-EMSE-LSU/Documentation/Notebook/July">Go to July Notebook</a></li> | ||
</ul> | </ul> | ||
</div> | </div> | ||
</html> | </html> | ||
Latest revision as of 01:06, 5 October 2013







{kind=link}