 "
"
Team:SYSU-China/Project/Design
From 2013.igem.org
(Created page with "<html> <head> <link rel="stylesheet" href="https://2013.igem.org/Team:SYSU-China/main.css?action=raw&ctype=text/css" type="text/css" /> <link rel="stylesheet" href="http://2013.ig...") |
|||
| (143 intermediate revisions not shown) | |||
| Line 2: | Line 2: | ||
<head> | <head> | ||
<link rel="stylesheet" href="https://2013.igem.org/Team:SYSU-China/main.css?action=raw&ctype=text/css" type="text/css" /> | <link rel="stylesheet" href="https://2013.igem.org/Team:SYSU-China/main.css?action=raw&ctype=text/css" type="text/css" /> | ||
| - | <link rel="stylesheet" href="https://2013.igem.org/Team:SYSU-China/Project_style.css?action=raw&ctype=text/css" type="text/css" /> | + | |
| + | <style type="text/css"> | ||
| + | body {background-color:#aaab4d;} | ||
| + | #cont_column h1,#cont_column h2,#cont_column h3 {color: #81822f;} | ||
| + | #cont_column h3 {border-bottom: 2px solid #aaab4d;} | ||
| + | #cont_column p,#cont_column li {color: #1d1d02;} | ||
| + | table {color:#1d1d02;background-color:transparent;} | ||
| + | </style> | ||
| + | |||
| + | <link rel="stylesheet" [[http://www.example.com link title][[http://www.example.com link title]]]href="https://2013.igem.org/Team:SYSU-China/Project_style.css?action=raw&ctype=text/css" type="text/css" /> | ||
<link rel="stylesheet" href="https://2013.igem.org/Team:SYSU-China/common_all.css?action=raw&ctype=text/css" type="text/css" /> | <link rel="stylesheet" href="https://2013.igem.org/Team:SYSU-China/common_all.css?action=raw&ctype=text/css" type="text/css" /> | ||
<script src="//ajax.googleapis.com/ajax/libs/jquery/1.10/jquery.min.js"></script> | <script src="//ajax.googleapis.com/ajax/libs/jquery/1.10/jquery.min.js"></script> | ||
| - | |||
| - | |||
| + | <script src="https://2013.igem.org/Team:SYSU-China/common.js?action=raw&ctype=text/javascript" type="text/javascript"></script> | ||
| + | |||
| + | <script src="https://2013.igem.org/Team:SYSU-China/animat.js?action=raw&ctype=text/javascript" type="text/javascript"></script> | ||
| + | |||
| + | <script src="https://2013.igem.org/Team:SYSU-China/Project.js?action=raw&ctype=text/javascript" type="text/javascript"></script> | ||
| + | |||
</head> | </head> | ||
<body> | <body> | ||
| + | <div id="outerdiv" style="position:fixed;top:0;left:0;background:rgba(220,220,220,0.9);z-index:2;width:100%;height:100%;display:none;"><div id="innerdiv" style="position:absolute;"><img id="bigimg" style="border:5px solid #fff;" src="" /></div></div> | ||
| + | |||
<DIV id="animat"> | <DIV id="animat"> | ||
<span><p>ipsc</p> | <span><p>ipsc</p> | ||
| Line 16: | Line 31: | ||
<DIV class="content_body" align="center"> | <DIV class="content_body" align="center"> | ||
<DIV class="navigater"> | <DIV class="navigater"> | ||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
</DIV> | </DIV> | ||
<DIV id="cont_column"> | <DIV id="cont_column"> | ||
| - | <!-- | + | <!--正文部分开始--> |
<DIV class="chapter"> | <DIV class="chapter"> | ||
| - | <span> | + | <span>Project/Design</span> |
| - | < | + | |
| - | <h2> | + | <h1>Overview</h1> |
| - | < | + | <br /><img src=" https://static.igem.org/mediawiki/2013/8/82/Design-overview.png " width="700" /><br /> |
| + | |||
| + | <p>Generally speaking, what we want to achieve is this: During the iPSCs differentiation, if the cell successfully turns into the cell we want, everything is fine; But once the cell remains to be the teratoma or accidently turns into the cancer cell, with our device, the cell will automatically trigger cell death. | ||
| + | In our project, we choose hepatocyte, which is a kind of normal liver cell, as the cell model we want. Our design has three parts: Killer, Sensor, and Switch. Each part will be introduced more specifically in the following.</p> | ||
| + | |||
| + | <p><strong>Killer: Suicide gene</strong></p> | ||
| + | <p>The strategy of our device to safeguard normal cells is to kill the tumor cells and teratoma cells. Instead of using medicines, which may cause side effect to other normal cells, we decide to use an exogenous suicide gene to trigger cell death. </p> | ||
| + | |||
| + | <p><strong>Sensor: miRNA-122 binding sites </strong></p> | ||
| + | <p>miRNA122 is a liver-specific small RNA, one of whose function is to supress the gene expression by either degrading the mRNA or by blocking its translation. We created several miRNA 122 binding sites downstream of suicide gene, so all unwanted cells, which have low expression level of miRNA122, will undergo cell death. </p> | ||
| + | |||
| + | <p><strong>Switch: Tet off system</strong></p> | ||
| + | <p>We lastly introduce a switch called tet-off system in our project in order to accurately control our killing system at the right time. Tet-off system is switched by a small molecular compound called Dox. Since most eukaryotic promoter like CMV have been reported to be silenced during iPSCs differentiation, we finaly modified Tet-off system by replacing the promoter CMV with another mammalian strong promoter EF-1α. </p> | ||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | <h1>Killer: Suicide Gene</h1> | ||
| + | <h2>Why suicide gene</h2> | ||
| + | |||
| + | <p> | ||
| + | In our project, we try to build a circuit that can prevent the iPSC-differentiated tissues from tumor formation. Instead of using some medicines, what we want to achieve here is to make the tumor cells kill themselves. So we decided to use proper suicide gene to trigger cel death.</p> | ||
| + | |||
<p> | <p> | ||
| - | + | The ideal suicide gene that fits our demand here should be the gene that can successfully induce cell death without causing any harmful effect to other normal tissues. From recent reports and papers, we found out that there have been many genes reported to have important roles in mammalian cell apoptosis. After devouring all these papers, our final candidates of suicide gene are listed below: | |
</p> | </p> | ||
| - | <img src="https://static.igem.org/mediawiki/2013/ | + | <div class="figure"> |
| + | <img class="fig_img" width="700px" src=" https://static.igem.org/mediawiki/2013/b/b3/SYSU-Design-%E8%87%AA%E6%9D%80%E5%9F%BA%E5%9B%A0.png " /> | ||
| + | <p class="des" style="margin-top:0px;width:700px"><strong>Figure 5.</strong> Candidates of suicide genes. </p> | ||
| + | <div class="clear"></div></div> | ||
<p> | <p> | ||
| - | + | The introduction of each genes about its suicide mechanisms and the reasons why we choose them are as follows. | |
| - | </p> | + | </p> |
| - | < | + | <h2>Which suicide gene</h2> |
| - | <h3> | + | <h3>hBax & hBax mutant<a class="quote">[1]</a></h3> |
<p> | <p> | ||
| - | + | hBax is a member of the Bcl-2 related protein family from human. This Family contains pro-apoptotic and anti-apoptotic proteins, and the balance among them determines the cell survival. hBax is the pro-apoptotic protein. During apoptosis, hBax will be inserted into the mitochondrial outer membrane and form permeable channels, release pro-apoptotic signals, finally lead to apoptosis. The pathway of apoptosis is showed in Figure 1. | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
</p> | </p> | ||
| + | <div class="figure"> | ||
| + | <img class="fig_img" width="700px" src=" https://static.igem.org/mediawiki/2013/7/75/SYSU-HBax1.png | ||
| + | " /> | ||
| + | <p class="des" style="margin-top:0px;width:700px"><strong>Figure 1.</strong> Apoptosis pathway induced by hBax. </p> | ||
| + | <div class="clear"></div></div> | ||
| + | |||
| + | |||
<p> | <p> | ||
| - | + | hBax S184a is a mutant of hBax that can constantly insert into mitochondrial outer membrane, thus we guess that it may have a stronger apoptosis-induced effect than normal hBax. | |
</p> | </p> | ||
<p> | <p> | ||
| - | + | It have been reported that over expression of this gene can successfully induce apoptosis in both Hela cell line and HEK-293 cell line <a class="quote">[2]</a>. Due to its generality, we finally determined to use it as one of our suicide genes candidates. | |
</p> | </p> | ||
<p> | <p> | ||
| - | + | However, our experiments results finally showed to us that, when expressed in Hep G2 cell line, which is a typical hepatoma cell line, they can not induce observable apoptosis. However, although it can not successfully kill Hep G2 cell line, we figured out that the pathway is conserved in yeast <a class="quote">[3]</a> ,so we also tried to transfect the gene and its mutant in yeast and finally proved that the killing effect is observable and even dose-dependent (<A HREF="http://parts.igem.org/Part:BBa_K1061006">BBa_K1061006</A>). So this extra result may be useful for other team in the future who want to apply safety device in yeast. We finally submitted the hBax as a biobrick and its mutant form as an improvement of the pre-existing part of part registry. | |
</p> | </p> | ||
| + | <p></p> | ||
| + | |||
| + | <h3>caspase 3</h3> | ||
| + | |||
<p> | <p> | ||
| - | + | caspase 3 is the last downstream executer of apoptosis in most mammalian cells <a class="quote">[4]</a>. Almost every apoptosis process needs the execution of caspase 3. As a cysteine protease, it can directly cleavage proteins inside cells and take part in DNA fragmentation. Caspase 3 contains two subunits, p17 and p12, which are translated in the same ORF. When cleavaged by caspase 9, another kind of protease involving in apoptosis, they will form a dimer that will act as an active form. | |
</p> | </p> | ||
| - | |||
| - | |||
<p> | <p> | ||
| - | + | Actually we split its gene into two parts, p17 and p12, and we used leuzine zipper to direct the dimerization of the two subunits<a class="quote">[5]</a>. | |
</p> | </p> | ||
| + | <div class="figure"> | ||
| + | <img class="fig_img" width="700px" src=" https://static.igem.org/mediawiki/2013/8/84/SYSU-Caspase.png " /> | ||
| + | <p class="des" style="margin-top:0px;width:700px"><strong>Figure 2.</strong> Mechanism of caspase apoptosis pathway </p> | ||
| + | <div class="clear"></div></div> | ||
| + | |||
| + | <div class="figure"> | ||
| + | <img class="fig_img" width="700px" src=" https://static.igem.org/mediawiki/2013/5/5f/SYSU-Caspase2.png " /> | ||
| + | <p class="des" style="margin-top:0px;width:700px"><strong>Figure 3. </strong> Combination of p17 and p12 </p> | ||
| + | <div class="clear"></div></div> | ||
| + | |||
<p> | <p> | ||
| - | + | However, in our project we finally decided to drop trying this gene, mainly for 3 reasons: | |
</p> | </p> | ||
| + | <ol> | ||
| + | <li>The apoptotic effect needs two subunit to be expressed simultaneously, which would increase the complication of our circuit.</li> | ||
| + | <li>To overcome the anti-apoptotic protein XIAP, which is high-expressed in Hep G2 cell line<a class="quote">[6]</a>, we may need an extremely high expression of caspase 3, which would also be a problem for our experiment.</li> | ||
| + | <li>We have mistakenly clone the wrong ORF of two subunits from the plasmid, so it leave us no time to do the experiment of caspase 3 before regional jamboree=.=.</li> | ||
| + | </ol> | ||
| + | |||
<p> | <p> | ||
| - | + | Besides, we may try to test another version of active caspase 3---the reconstitute caspase 3 in following days<a class="quote">[7]</a>. | |
</p> | </p> | ||
| + | |||
| + | <h3>rip 1</h3> | ||
<p> | <p> | ||
| - | + | rip1 is the abbreviation of Receptor Interacting Protein kinase 1 in mammalian cells. It is an important regulator of cell survival and death, taking part in several program cell death pathways. It has been reported that over expression of rip 1 can induce both apoptosis and necrosis in certain cell lines. <a class="quote">[8]</a> So it is a potential suitable suicide gene that we can use in our device.</p> | |
| + | <div class="figure"> | ||
| + | <img class="fig_img" width="700px" src=" https://static.igem.org/mediawiki/2013/2/22/SYSU-Rip1.png " /> | ||
| + | <p class="des" style="margin-top:0px;width:700px"><strong>Figure 4. </strong> Pathway of rip1 </p> | ||
| + | <div class="clear"></div></div> | ||
| + | |||
| + | <p> | ||
| + | We tested rip1 in several cell lines, including HTC-75, Bosc, and Hep G2 cell lines. What we have observed is a mix of both apoptosis and necrosis. Although it can induce necrosis in cancer cell lines, which may potentially cause inflammation in tissues, we still consider it as our choice of suicide gene. The reasons are as follows: | ||
</p> | </p> | ||
| + | |||
<p> | <p> | ||
| - | + | <ol> | |
| + | <li>We know that in iPSC differentiation period, only a small fraction of cells will become cancerous at a certain time, so the necrosis of these cells would not likely to lead to severe inflammation.</li> | ||
| + | <li>Many apoptosis pathways have been reported to be blocked in tumor cells, but there is some evidence revealing that when the apoptosis pathways are blocked the necrosis pathway will be activated. Besides, some recent papers showed that rip1 can trigger cell necrosis only with extra exogenous medicine. In other circumstances rip1 could lead to apoptosis.</li> | ||
| + | <li>We have cloned another receptor interacting protein kinase, rip 3, which has been proved to interact with rip 1 and lead to necrosis, so we think that even the over expression of rip 1 can not successfully kill the cells, it may also co-express with rip 3.</li> | ||
| + | </ol> | ||
</p> | </p> | ||
| - | < | + | <p> |
| - | + | Surely, we will keep trying to find genes that only lead to apoptosis but not necrosis in the future, the better solution, in our mind, may be a combination of multiple apoptotic genes. | |
| - | + | </p> | |
| - | + | ||
| - | + | <h3>rip 3</h3> | |
| - | + | ||
| - | + | ||
| - | + | <p> | |
| - | + | rip3, like rip 1, is also a member of Receptor Interacting Protein family. It works via the interaction with rip 1, and thus induces the necrosis pathway which rip 1 mediated. <a class="quote">[9]</a> | |
| - | + | </p> | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
<p> | <p> | ||
| - | + | However, it have also been reported that over expression of rip 3 in certain cell lines can induce apoptosis.In our experiment, we found out that rip 3 can lead to cell death in several cell lines, including HTC-75 cell lines and HEK-293 cell lines. | |
</p> | </p> | ||
<p> | <p> | ||
| - | + | Because rip 3 mainly induces necrosis, we select it as our candidate of suicide genes for the same reasons as we select rip 1. And we also suggest that when using rip 3 to cure cancer, tight control expression system may be necessary.</p> | |
| + | |||
| + | <h3>apoptin <a class="quote">[10]</a></h3> | ||
| + | |||
| + | <p> | ||
| + | apoptin, a protein first isolated from Chicken anemia virus, has been regarded as a potential drug for cancer treatment. It has been reported in over 70 cell lines that apoptin can selectively kill cancer cells but not normal cells. The result of in vivo test in mice is very exciting: the intraperitoneal injection of vector carrying the apoptin seems not confer any observable side effect on mice. | ||
</p> | </p> | ||
| + | <p> | ||
| + | The mechanism of how apoptin works is not fully understood. It probably works via the non-p53 apoptosis pathway,hence is not easy to be blocked. The localization of apoptin in cancer cells is in nuclear, while in normal cells it locates in cytoplasm . This was proved by our experiment results.</p> | ||
| + | |||
| + | <p> | ||
| + | A special character of apoptin is that it works like a sensor, probably by recognizing certain early signals of cancer formation. These signals may be general, which explained the reason why the apoptin can kill such a broad spectrum of cancer cells. So the construct of EF-1α-apoptin may provide a general circuit for safety issues of gene therapy and renew the concept of sensor. Making use of the by-stander effect, TAT-apoptin or SP-TAT-apoptin <a class="quote">[11]</a>may be powerful and provide a "safe environment" for the cells under genetic manipulation. | ||
| + | </p> | ||
| + | |||
| + | <p> | ||
| + | However, we still have to say that although apoptin has been proved to be safe for many normal cell types, it has not been proved in all normal cell types. So we should consider and test thoroughly about the immune reaction effect, making sure that it does not happen or can be controlled. | ||
| + | </p> | ||
| + | |||
| + | |||
| + | <h2>Future work: Combination of suicide genes? Or using the scalable miRNA circuit? </h2> | ||
| + | |||
| + | <p> | ||
| + | With our project proceeded, we found out that apoptosis pathways are generally blocked in cancer cell lines (maybe this's one of the reasons why cancers are so hard to deal with). However, Synthetic biology has a great advantage in designing complicated circuits, and hence, it will be possible to combine several suitable suicide gene together and construct a circuit that can overcome the blocking problem of apoptosis pathway in cancer cells. And by screening the publication work we also found out that the shRNA and miRNA expression technique, which is controllable <a class="quote">[12]</a>, can successfully knock-down gene expression. | ||
| + | </p> | ||
| + | <p> | ||
| + | The circuit design based on shRNA and miRNA should be scalable, because the sequence of them are short enough so that the whole circuit can be much more complicated than the design base on suicide proteins. The broader target of miRNA and shRNA is, the harder to be blocked. For this reason, the short RNA -based circuit, including CRISPRi, may be a promising way for cancer treatment and gene therapy design. | ||
| + | </p> | ||
| + | |||
| + | |||
| + | <h1>Sensor: miR-122 binding sites</h1> | ||
| + | <h2> Why miRNA</h2> | ||
| + | <p> | ||
| + | Now we have effective suicide gene for our system. To achieve the selective killing, we want to find a sensor that can distinguish the wanted and unwanted cells,so the task falls on finding the distinguishing molecular markers. In our selective system,only somatic cells are supposed to survive. However, as there are too many types of somatic cells, it will be too difficult to save them all with only one device, we decided to choose one or several types of somatic cell as our model. | ||
| + | We come up with two strategies: tissue-specific activation and tissue-specific repression. At the begining, we want to find tissue-specific promoters but after massive search,we realized that such promoters cannot be universally activated in all kinds of cancer cells. So we then move to think about using suppressor- miRNA. MicroRNAs can regulate gene expression and control many important process including development, differentiation, apoptosis and proliferation. They regulate gene expression by binding to a short 6 nt region within the 3'UTR of their target mRNAs. After screening the expression map of all kinds of miRNA in different tissues and organs and found out that, there is a small RNA called mircoRNA122 has a specificly high expreesion in the liver cells. | ||
| + | |||
| + | </p> | ||
| + | <p> | ||
| + | |||
| + | <h2> Which miRNA</h2> | ||
| + | |||
| + | <p> | ||
| + | MicroRNA-122(miR-122) is a liver-specific microRNA<a class="quote">[13]</a> that is most abundantly expressed in the liver as it accounts for approximately 70% of all hepatic icroRNAs. Interestingly, it is specifically highly expressed in normal hepatocytes instead of undifferentiated or cancerous cells. According to this, we have designed a complementary miR-122 biding site (sometimes we prefer to call it miR122 traget) as a sensor to select the healthy hepatocytes we want when high level of endogenous miR-122 bind to our target in 3'UTR and then degrade the mRNA of suicide gene. That is to say, during direction differentiation, undifferentiated or wrong kinds of cells will express the suicide gene because they have low level of miR-122. iPS-derived hepatocytes do not express deadly level of suicide gene unless it becomes cancer cells soontaneously. | ||
| + | </p> | ||
| + | <div class="figure"> | ||
| + | <img class="fig_img" width="700px" src=" https://static.igem.org/mediawiki/2013/2/2e/SYSU-Design-122%E8%A1%A8%E8%BE%BE.png | ||
| + | " /> | ||
| + | <p class="des" style="margin-top:0px;width:700px"><strong>Figure 1.</strong> Expression of miRNA122 in differerent hepa cell lines. </p> | ||
| + | <div class="clear"></div></div> | ||
| + | <div class="figure"> | ||
| + | <img class="fig_img" width="700px" src=" https://static.igem.org/mediawiki/2013/7/71/SYSU-Design-122%E5%8E%9F%E7%90%86.png " /> | ||
| + | <p class="des" style="margin-top:0px;width:700px"><strong>Figure 2.</strong> The principle of miRNA </p> | ||
| + | <div class="clear"></div></div> | ||
| + | |||
| + | <p> | ||
| + | For more information please go to the background page. (<A HREF="https://2013.igem.org/Team:SYSU-China/Project?#h2_0">Background</A>) | ||
| + | |||
| + | <h2>Why liver</h2> | ||
| + | <p> | ||
| + | Liver cancer is the fifth deadly cancer in the world and the situation is more serious in China as 55% of the patients are from China. Every year, thousands of patients wait for liver transplant to save their lives. Recent advances in iPS-derived hepatocytes<a class="quote">[14]</a> provides an easier and more convenient way to solve the problem of donor shortage and reduce immune reaction. iPS-derived hepatocytes are also required for scanning for hepatic toxicity with the advantages of batch to batch stability and more resources. What's more, patients with inherent metabolic diseases can benefit from iPS-derived hepatocytes with genetic information modified. Here, we choose the iPS-derived hepatocytes as a model to test our idea. We have selected the liver-specific miR-122 as a marker to distinguish normal, differentiated liver cells from undifferentiated or cancerous cells. Our device provides a solution to increase the efficiency of directional differentiation and prevent possible carcinogenesis. | ||
| + | </p> | ||
| + | <p> | ||
| + | For more information, please go to the background page. | ||
| + | </p> | ||
| + | |||
| + | <h1> Switch: Tet-off system</h1> | ||
| + | |||
| + | <h2>Why switch </h2> | ||
| + | |||
| + | <p> | ||
| + | Considering liver cell is very resistant to any transfection or stable infection, our device is supposed to be delivered into cells before iPSC reprograming period. That means, we infect our device along with four transcriptional facts into somatic cells by lentivirus and then induse them into iPSCs. In order to not kill iPSCs immediately, given that the miRNA122 level is very low, we want to add a manual switch to accurately control our killing system at the right time. | ||
| + | </p> | ||
| + | <p> | ||
| + | Tet control system is a beautiful combination of prokaryotic and eukaryotic gene-expression regulatory system. It consists of two parts: the regulatory protein tTA or rtTA and a downstream response element. TetR is a repressor from E. coli, which can block the downstream expression when it binds to a TetO operon. When tetracycline(Tc) or doxycycline(Dox) is absent, gene expression is repressed and when Tc or Dox is introduced to the medium, it leaves the TetO site and let the gene of interest express. The whole tTA regulatory protein is fused by TetR and an activation domain VP16, which will still bind to TetO when dox is absence. However, by alternating four amino acids of tTA, which called rtTA, it binds to tetO operon when Dox is present. When the 7 consecutive tetO sequences are fused with a minimal CMV promoter, we will get a regulatory promote, TRE, that is activated when tTA or rtTA binds to it. | ||
| + | </p> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/3/37/SYSU-Tet_on.png" width="700" /> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/4/48/SYSU-Tet-off.png" width="700" /> | ||
| + | <p> | ||
| + | |||
| + | |||
| + | <h2>Which Tet system</h2> | ||
| + | <h3>On or off, this is a question.</h3> | ||
| + | <p> | ||
| + | Basically, there are two kinds of tet control system. The first one, tet-off, is based on tTA, which will activate downstream expression when Tc or Dox is absence. The second one, tet-on, is based on rtTA, which will activate downstream expression when Dox is added.</p> | ||
| + | <p> | ||
| + | So, which one should we use? | ||
| + | </p> | ||
| + | <p> | ||
| + | Our construction need to express the suicide gene when detecting the cells are cancerous. So the tet part should be activated in vivo, only suppressed by the miRNA-122. After the transplantation, it will be inconvenient to intake dox constantly, as dox is a kind of antibiotics. Also, To intake dox after transplantation will need higher dose of dox to achieve the same level of expression, comparing to add dox into culture medium. So, the better choice will be using tet-off system, which can achieve constantly expression without adding dox. For preventing the expression of suicide gene in iPS cells, we only need to add dox during the in-vitro differentiation. | ||
| + | </p> | ||
| + | <p> | ||
| + | However, combining a special suicide gene apoptin, which will not kill normal cells when constantly expressing, with another suicide gene, it is possible to use tet-on system without constantly intake the dox in vivo. And the time of adding dox will be shorter. | ||
| + | </p> | ||
| + | |||
| + | <h3>One, two, three: which is the golden generation?</h3> | ||
| + | |||
| + | <p> | ||
| + | There are three generations of tet-expression systems now <a class="quote">[15]</a>. | ||
| + | |||
| + | </p> | ||
| + | <p> | ||
| + | We expect that our suicide gene has low level of leakage expression and a high fold increasement. | ||
| + | First, we want to find out the one with lowest leakage expression. It turns out to be tTA+p_tight. However, a low leakage expression is usually in accompany with low maximal expression. Hence, transient transfection proved that the suicide gene is not strong enough to kill all cells. But cells escaped from surveillance might go through rapid genetic rearrangement to acquire invasiveness. | ||
| + | Actually we have gotten another type of Tet-off system fused with KRAB transcription factor <a class="quote">[16]</a>. It has a higher leakage expression but the fold increasement is the best. Actually, this problem can be circumvented in the process of generating stable cell lines because leakage expression differs from clone to clone and is lower than in transient transfection. However, KRAB does not function in ES cells and this Tet-off is abandoned. | ||
| + | We are also interested in alternative Tet system like those controlling the expression of RNA and the else. | ||
| + | |||
| + | <div class="figure"> | ||
| + | <img class="fig_img" width="700px" src=" https://static.igem.org/mediawiki/2013/a/a5/SYSU-New_tet.png " /> | ||
| + | <p class="des" style="margin-top:0px;width:700px"><strong>Figure 7. </strong> Mechanism of a new tet system. </p> | ||
| + | <div class="clear"></div></div> | ||
| + | |||
| + | |||
| + | <h2>pEF-1α or pCMV</h2> | ||
| + | <p> | ||
| + | To consistently express the protein tTA of our circuit in a high level, we finally select a human elongation factor 1 alpha ( PEF-1α ) to replace the original promoter pCMV to be our promoter in tet-off system. | ||
| + | </p> | ||
| + | <p> | ||
| + | <h3>Generous & strong</h3> | ||
| + | <p> | ||
| + | Since Elongation Factor 1α takes part in translation, almost every types of mammalian cells need to express this factor. Hence its promoter, EF-1alpha, is constantly activated no matter what kind of state the cell is undergoing <a class="quote">[17]</a>. In our design, the expression of our device is supposed to be stable and persistent during the whole differentiating process, so we choose EF-1alpha promoter to drive tTA transcription. Besides, EF-1alpha promoter is one of the strongest promoters that has been used in constructing mammalian circuit. | ||
| + | </p> | ||
| + | <p> | ||
| + | <h3>No silence in PSCs</h3> | ||
| + | <p> | ||
| + | EF-1alpha promoter has been proved to be particularly useful in constructing stable cell lines. Compared to another strong promoter, CMV promoter, which has also been widely used in constructing mammalian circuit, EF-1alpha promoter won't undergo transgenic silence in certain cell types, including the iPS cells in our project and hematopoietic stem cells et al <a class="quote">[18]</a>. | ||
| + | </p> | ||
| + | <p> | ||
| + | Previous work has showed that the expression level of gene driven by EF-1alpha promoter is more robust in several cell lines, which will provide convenience to construct general circuit used in different cell lines. The data that characterize in one type of cell lines will be reliable in other types. | ||
| + | </p> | ||
| + | <h1>lentivrus </h1> | ||
| + | <p> | ||
| + | We decided to use lentivirus vector to carry our circuits, deliver and long-term maintain of our circuit in our chassis genome. We will introduce the mechanism of lentiviral transfection system and the reasons why we choose it. | ||
| + | </p> | ||
| + | <h2>What is lentivirus</h2> | ||
| + | <p> | ||
| + | Lentiviral vectors (LV) are viral-based gene delivery systems that can stably deliver genes or RNAi into primary cells or cell lines with up to 100% efficiency. LVs bind to target cells using an envelope protein which allows for release of the LV RNA containing the gene or gene silencing sequence into the cell. The LVs RNA is then converted into DNA using an enzyme called reverse transcriptase by a process called reverse transcription. The DNA pre-integration complex then enters the nucleus and integrates into the target cell's chromosomal DNA. Gene delivery is stable because the target gene or gene silencing sequence is integrated in the chromosome and is copied along with the DNA of the cell every time the cell divides. One of the discriminating features of LVs is their ability to integrate into non-dividing cells, in contrast to other vectors that either don't integrate efficiently into chromosomal DNA (e.g. non-viral, Adenoviral and Adenoviral-Associated vectors) or can only integrate upon cell division (e.g. conventional Retroviral vectors). | ||
| + | </p> | ||
| + | |||
| + | <h2>Why lentivirus</h2> | ||
| + | <p> | ||
| + | Because our device is supposed to be expressed stably in a long term and therefore persistently safeguard the cells under any condition, we finally choose lentivirus to be our delivery system into iPSCs, for its high transfection efficiency and the ability to integration into host genome. Though we can never escape the fact that the use of lentivirus could increase the oncogenic risks in cells at the same time, however, considering another fact that almost every inducing/reprogramming factors in iPSCs are oncogenes, maybe the better thing we could do here is to deliver a monitor device which would persistently safeguard the cells in a long term. | ||
| + | </p> | ||
| + | <p> | ||
| + | To sum up, we choose lentivirus as our delivery system mainly for the following reasons: | ||
| + | <ul> | ||
| + | <li>Lentivector can integrate into human genome in a multi-copy way.</li> | ||
| + | <li>Lentivector can stably expressed in wide types of cell lines, without being silenced or irreversible transgenic suppression in ESCs and iPSCs.</li> | ||
| + | <li>Lentivector has a relatively higher efficiency compared with other integrating virus.</li> | ||
| + | <li>Lentivector is relatively safer in all integrating virus. </li> | ||
| + | </ul></p> | ||
| + | <p> | ||
| + | The lentivector we used in our project is the 3rd generation inducible lentivirus pPRIME, whose packaging plasmids are psPAX2 and pMD2G. We packaged lentivirus mix in 293T cell lines. | ||
| + | </p> | ||
| + | |||
| + | <h1>All parts together! </h1> | ||
| + | <p>Up till now, we have introduced all the gene parts. Combining them toghther, our cuicurt finally works like this:</p> | ||
| + | <div class="figure"> | ||
| + | <img class="fig_img" width="700px" src=" https://static.igem.org/mediawiki/2013/5/56/SYSU-Design-all.png " /> | ||
| + | <p class="des" style="margin-top:0px;width:700px"><strong>Figure 8.</strong> Mechanism of our final curcuit. iPSCs are firstly cultured with Dox. Once we want to turn on our device, Dox will be removed,after which the expression level of endogenous miRNA122 will be the only key. When iPSCs are successfully differentiated into hepatocytes, high level of miRNA122 will bind to the targets and therefore slience suicide mRNA tranlation. However, when tumorigenesis happens in the cell, the level of miRNA122 drop significantly, thus the suicide protein will finally lead to cell apopotosis. Therefore, those tumor cells are picked out from the iPSC mass. </p> | ||
| + | <div class="clear"></div></div> | ||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
<DIV id="references"> | <DIV id="references"> | ||
<h2>References</h2> | <h2>References</h2> | ||
| - | <p><a class="references">[1]</a>The | + | |
| - | <p><a class="references">[2]</a> | + | |
| - | <p><a class="references">[3]</a> | + | <p><a class="references">[1]</a> Amotz Nechushtan et al, The EMBO Journal,18,(1999)</p> |
| - | <p><a class="references">[4]</a> | + | <p><a class="references">[2]</a> Zhen Xie et al.,science, Science 333, 1307 (2011)</p> |
| - | <p><a class="references">[5]</a> | + | <p><a class="references">[3]</a> Qunli Xu et al,molecular cell, 1,3, 1998</p> |
| - | <p><a class="references">[6]</a> | + | <p><a class="references">[4]</a> ]AG Porter et al,cell death and differentiation,6,2,(1999)</p> |
| - | <p><a class="references">[7]</a> | + | <p><a class="references">[5]</a> Dattananda S. Chelur et al,PNAS,104, 7,2007</p> |
| - | <p><a class="references">[8]</a> | + | <p><a class="references">[6]</a> Xuanyong Lu etal, Int. J. Med. Sci.,2,1 ,(2005)</p> |
| - | <p><a class="references">[9]</a> | + | <p><a class="references">[7]</a> Srinivasa M. Srinivasula et al,, J. Biol. Chem,273,10107,(1997)</p> |
| - | <p><a class="references">[10]</a> | + | <p><a class="references">[8]</a> N Festjens,Cell death and differentiation,14,400,(2007)</p> |
| - | <p><a class="references">[11]</a> | + | <p><a class="references">[9]</a> Liming Sun et al,Cell, 148,213,2012</p> |
| - | <p><a class="references">[12]</a> | + | <p><a class="references">[10]</a> Liming Sun et al,Cell, 148,213,2012</p> |
| - | <p><a class="references">[ | + | <p><a class="references">[11]</a> Su-Xia Han et al,14,23,(2008)</p> |
| + | <p><a class="references">[12]</a>http://www.clontech.com/CN/Products/Cell_Biology_and_Epigenetics/RNA_Interference/MicroRNA/Tet-Inducible_MicroRNA?sitex=10022:22372:US</p> | ||
| + | <p><a class="references">[13]</a> Hui Xu et al,Hepatology,52,4,(2010)</p> | ||
| + | <p><a class="references">[14]</a> Takanori Takebe et al, Nature,499,481(2013)</p> | ||
| + | <p><a class="references">[15]</a> http://www.clontech.com/CN/Products/Inducible_Systems/Tetracycline-Inducible_Expression/Tet-On_3G?sitex=10022:22372:US</p> | ||
| + | <p><a class="references">[16]</a> Jolanta Szulc et al,nature method, 3,2 (2006)</p> | ||
| + | <p><a class="references">[17]</a> Negrutskii BS, Prog Nucleic Acid Res Mol Biol.,60,47,(1998)</p> | ||
| + | <p><a class="references">[18]</a>http://www.clontech.com/CN/Products/Inducible_Systems/Tetracycline-Inducible_Expression/Tet-On_3G_EF1-Alpha_Promoter?sitex=10022:22372:US</p> | ||
| + | |||
| + | |||
| + | |||
| + | </DIV> | ||
</DIV> | </DIV> | ||
Latest revision as of 03:53, 29 October 2013
ipsc

Overview

Generally speaking, what we want to achieve is this: During the iPSCs differentiation, if the cell successfully turns into the cell we want, everything is fine; But once the cell remains to be the teratoma or accidently turns into the cancer cell, with our device, the cell will automatically trigger cell death. In our project, we choose hepatocyte, which is a kind of normal liver cell, as the cell model we want. Our design has three parts: Killer, Sensor, and Switch. Each part will be introduced more specifically in the following.
Killer: Suicide gene
The strategy of our device to safeguard normal cells is to kill the tumor cells and teratoma cells. Instead of using medicines, which may cause side effect to other normal cells, we decide to use an exogenous suicide gene to trigger cell death.
Sensor: miRNA-122 binding sites
miRNA122 is a liver-specific small RNA, one of whose function is to supress the gene expression by either degrading the mRNA or by blocking its translation. We created several miRNA 122 binding sites downstream of suicide gene, so all unwanted cells, which have low expression level of miRNA122, will undergo cell death.
Switch: Tet off system
We lastly introduce a switch called tet-off system in our project in order to accurately control our killing system at the right time. Tet-off system is switched by a small molecular compound called Dox. Since most eukaryotic promoter like CMV have been reported to be silenced during iPSCs differentiation, we finaly modified Tet-off system by replacing the promoter CMV with another mammalian strong promoter EF-1α.
Killer: Suicide Gene
Why suicide gene
In our project, we try to build a circuit that can prevent the iPSC-differentiated tissues from tumor formation. Instead of using some medicines, what we want to achieve here is to make the tumor cells kill themselves. So we decided to use proper suicide gene to trigger cel death.
The ideal suicide gene that fits our demand here should be the gene that can successfully induce cell death without causing any harmful effect to other normal tissues. From recent reports and papers, we found out that there have been many genes reported to have important roles in mammalian cell apoptosis. After devouring all these papers, our final candidates of suicide gene are listed below:

Figure 5. Candidates of suicide genes.
The introduction of each genes about its suicide mechanisms and the reasons why we choose them are as follows.
Which suicide gene
hBax & hBax mutant[1]
hBax is a member of the Bcl-2 related protein family from human. This Family contains pro-apoptotic and anti-apoptotic proteins, and the balance among them determines the cell survival. hBax is the pro-apoptotic protein. During apoptosis, hBax will be inserted into the mitochondrial outer membrane and form permeable channels, release pro-apoptotic signals, finally lead to apoptosis. The pathway of apoptosis is showed in Figure 1.

Figure 1. Apoptosis pathway induced by hBax.
hBax S184a is a mutant of hBax that can constantly insert into mitochondrial outer membrane, thus we guess that it may have a stronger apoptosis-induced effect than normal hBax.
It have been reported that over expression of this gene can successfully induce apoptosis in both Hela cell line and HEK-293 cell line [2]. Due to its generality, we finally determined to use it as one of our suicide genes candidates.
However, our experiments results finally showed to us that, when expressed in Hep G2 cell line, which is a typical hepatoma cell line, they can not induce observable apoptosis. However, although it can not successfully kill Hep G2 cell line, we figured out that the pathway is conserved in yeast [3] ,so we also tried to transfect the gene and its mutant in yeast and finally proved that the killing effect is observable and even dose-dependent (BBa_K1061006). So this extra result may be useful for other team in the future who want to apply safety device in yeast. We finally submitted the hBax as a biobrick and its mutant form as an improvement of the pre-existing part of part registry.
caspase 3
caspase 3 is the last downstream executer of apoptosis in most mammalian cells [4]. Almost every apoptosis process needs the execution of caspase 3. As a cysteine protease, it can directly cleavage proteins inside cells and take part in DNA fragmentation. Caspase 3 contains two subunits, p17 and p12, which are translated in the same ORF. When cleavaged by caspase 9, another kind of protease involving in apoptosis, they will form a dimer that will act as an active form.
Actually we split its gene into two parts, p17 and p12, and we used leuzine zipper to direct the dimerization of the two subunits[5].

Figure 2. Mechanism of caspase apoptosis pathway

Figure 3. Combination of p17 and p12
However, in our project we finally decided to drop trying this gene, mainly for 3 reasons:
- The apoptotic effect needs two subunit to be expressed simultaneously, which would increase the complication of our circuit.
- To overcome the anti-apoptotic protein XIAP, which is high-expressed in Hep G2 cell line[6], we may need an extremely high expression of caspase 3, which would also be a problem for our experiment.
- We have mistakenly clone the wrong ORF of two subunits from the plasmid, so it leave us no time to do the experiment of caspase 3 before regional jamboree=.=.
Besides, we may try to test another version of active caspase 3---the reconstitute caspase 3 in following days[7].
rip 1
rip1 is the abbreviation of Receptor Interacting Protein kinase 1 in mammalian cells. It is an important regulator of cell survival and death, taking part in several program cell death pathways. It has been reported that over expression of rip 1 can induce both apoptosis and necrosis in certain cell lines. [8] So it is a potential suitable suicide gene that we can use in our device.

Figure 4. Pathway of rip1
We tested rip1 in several cell lines, including HTC-75, Bosc, and Hep G2 cell lines. What we have observed is a mix of both apoptosis and necrosis. Although it can induce necrosis in cancer cell lines, which may potentially cause inflammation in tissues, we still consider it as our choice of suicide gene. The reasons are as follows:
- We know that in iPSC differentiation period, only a small fraction of cells will become cancerous at a certain time, so the necrosis of these cells would not likely to lead to severe inflammation.
- Many apoptosis pathways have been reported to be blocked in tumor cells, but there is some evidence revealing that when the apoptosis pathways are blocked the necrosis pathway will be activated. Besides, some recent papers showed that rip1 can trigger cell necrosis only with extra exogenous medicine. In other circumstances rip1 could lead to apoptosis.
- We have cloned another receptor interacting protein kinase, rip 3, which has been proved to interact with rip 1 and lead to necrosis, so we think that even the over expression of rip 1 can not successfully kill the cells, it may also co-express with rip 3.
Surely, we will keep trying to find genes that only lead to apoptosis but not necrosis in the future, the better solution, in our mind, may be a combination of multiple apoptotic genes.
rip 3
rip3, like rip 1, is also a member of Receptor Interacting Protein family. It works via the interaction with rip 1, and thus induces the necrosis pathway which rip 1 mediated. [9]
However, it have also been reported that over expression of rip 3 in certain cell lines can induce apoptosis.In our experiment, we found out that rip 3 can lead to cell death in several cell lines, including HTC-75 cell lines and HEK-293 cell lines.
Because rip 3 mainly induces necrosis, we select it as our candidate of suicide genes for the same reasons as we select rip 1. And we also suggest that when using rip 3 to cure cancer, tight control expression system may be necessary.
apoptin [10]
apoptin, a protein first isolated from Chicken anemia virus, has been regarded as a potential drug for cancer treatment. It has been reported in over 70 cell lines that apoptin can selectively kill cancer cells but not normal cells. The result of in vivo test in mice is very exciting: the intraperitoneal injection of vector carrying the apoptin seems not confer any observable side effect on mice.
The mechanism of how apoptin works is not fully understood. It probably works via the non-p53 apoptosis pathway,hence is not easy to be blocked. The localization of apoptin in cancer cells is in nuclear, while in normal cells it locates in cytoplasm . This was proved by our experiment results.
A special character of apoptin is that it works like a sensor, probably by recognizing certain early signals of cancer formation. These signals may be general, which explained the reason why the apoptin can kill such a broad spectrum of cancer cells. So the construct of EF-1α-apoptin may provide a general circuit for safety issues of gene therapy and renew the concept of sensor. Making use of the by-stander effect, TAT-apoptin or SP-TAT-apoptin [11]may be powerful and provide a "safe environment" for the cells under genetic manipulation.
However, we still have to say that although apoptin has been proved to be safe for many normal cell types, it has not been proved in all normal cell types. So we should consider and test thoroughly about the immune reaction effect, making sure that it does not happen or can be controlled.
Future work: Combination of suicide genes? Or using the scalable miRNA circuit?
With our project proceeded, we found out that apoptosis pathways are generally blocked in cancer cell lines (maybe this's one of the reasons why cancers are so hard to deal with). However, Synthetic biology has a great advantage in designing complicated circuits, and hence, it will be possible to combine several suitable suicide gene together and construct a circuit that can overcome the blocking problem of apoptosis pathway in cancer cells. And by screening the publication work we also found out that the shRNA and miRNA expression technique, which is controllable [12], can successfully knock-down gene expression.
The circuit design based on shRNA and miRNA should be scalable, because the sequence of them are short enough so that the whole circuit can be much more complicated than the design base on suicide proteins. The broader target of miRNA and shRNA is, the harder to be blocked. For this reason, the short RNA -based circuit, including CRISPRi, may be a promising way for cancer treatment and gene therapy design.
Sensor: miR-122 binding sites
Why miRNA
Now we have effective suicide gene for our system. To achieve the selective killing, we want to find a sensor that can distinguish the wanted and unwanted cells,so the task falls on finding the distinguishing molecular markers. In our selective system,only somatic cells are supposed to survive. However, as there are too many types of somatic cells, it will be too difficult to save them all with only one device, we decided to choose one or several types of somatic cell as our model. We come up with two strategies: tissue-specific activation and tissue-specific repression. At the begining, we want to find tissue-specific promoters but after massive search,we realized that such promoters cannot be universally activated in all kinds of cancer cells. So we then move to think about using suppressor- miRNA. MicroRNAs can regulate gene expression and control many important process including development, differentiation, apoptosis and proliferation. They regulate gene expression by binding to a short 6 nt region within the 3'UTR of their target mRNAs. After screening the expression map of all kinds of miRNA in different tissues and organs and found out that, there is a small RNA called mircoRNA122 has a specificly high expreesion in the liver cells.
Which miRNA
MicroRNA-122(miR-122) is a liver-specific microRNA[13] that is most abundantly expressed in the liver as it accounts for approximately 70% of all hepatic icroRNAs. Interestingly, it is specifically highly expressed in normal hepatocytes instead of undifferentiated or cancerous cells. According to this, we have designed a complementary miR-122 biding site (sometimes we prefer to call it miR122 traget) as a sensor to select the healthy hepatocytes we want when high level of endogenous miR-122 bind to our target in 3'UTR and then degrade the mRNA of suicide gene. That is to say, during direction differentiation, undifferentiated or wrong kinds of cells will express the suicide gene because they have low level of miR-122. iPS-derived hepatocytes do not express deadly level of suicide gene unless it becomes cancer cells soontaneously.

Figure 1. Expression of miRNA122 in differerent hepa cell lines.

Figure 2. The principle of miRNA
For more information please go to the background page. (Background)
Why liver
Liver cancer is the fifth deadly cancer in the world and the situation is more serious in China as 55% of the patients are from China. Every year, thousands of patients wait for liver transplant to save their lives. Recent advances in iPS-derived hepatocytes[14] provides an easier and more convenient way to solve the problem of donor shortage and reduce immune reaction. iPS-derived hepatocytes are also required for scanning for hepatic toxicity with the advantages of batch to batch stability and more resources. What's more, patients with inherent metabolic diseases can benefit from iPS-derived hepatocytes with genetic information modified. Here, we choose the iPS-derived hepatocytes as a model to test our idea. We have selected the liver-specific miR-122 as a marker to distinguish normal, differentiated liver cells from undifferentiated or cancerous cells. Our device provides a solution to increase the efficiency of directional differentiation and prevent possible carcinogenesis.
For more information, please go to the background page.
Switch: Tet-off system
Why switch
Considering liver cell is very resistant to any transfection or stable infection, our device is supposed to be delivered into cells before iPSC reprograming period. That means, we infect our device along with four transcriptional facts into somatic cells by lentivirus and then induse them into iPSCs. In order to not kill iPSCs immediately, given that the miRNA122 level is very low, we want to add a manual switch to accurately control our killing system at the right time.
Tet control system is a beautiful combination of prokaryotic and eukaryotic gene-expression regulatory system. It consists of two parts: the regulatory protein tTA or rtTA and a downstream response element. TetR is a repressor from E. coli, which can block the downstream expression when it binds to a TetO operon. When tetracycline(Tc) or doxycycline(Dox) is absent, gene expression is repressed and when Tc or Dox is introduced to the medium, it leaves the TetO site and let the gene of interest express. The whole tTA regulatory protein is fused by TetR and an activation domain VP16, which will still bind to TetO when dox is absence. However, by alternating four amino acids of tTA, which called rtTA, it binds to tetO operon when Dox is present. When the 7 consecutive tetO sequences are fused with a minimal CMV promoter, we will get a regulatory promote, TRE, that is activated when tTA or rtTA binds to it.

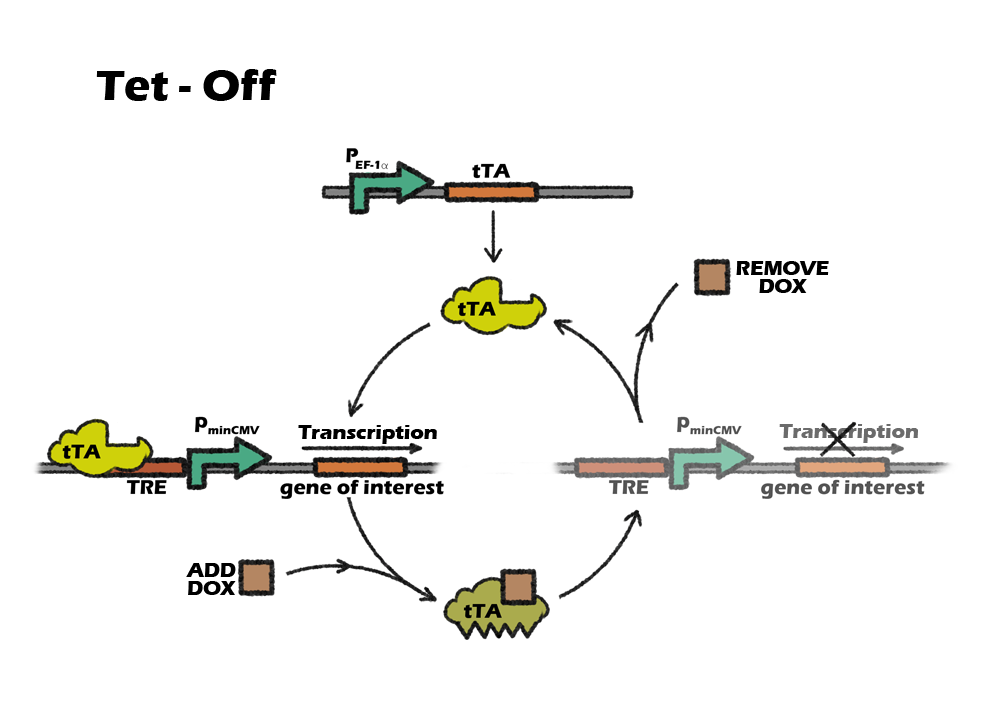
Which Tet system
On or off, this is a question.
Basically, there are two kinds of tet control system. The first one, tet-off, is based on tTA, which will activate downstream expression when Tc or Dox is absence. The second one, tet-on, is based on rtTA, which will activate downstream expression when Dox is added.
So, which one should we use?
Our construction need to express the suicide gene when detecting the cells are cancerous. So the tet part should be activated in vivo, only suppressed by the miRNA-122. After the transplantation, it will be inconvenient to intake dox constantly, as dox is a kind of antibiotics. Also, To intake dox after transplantation will need higher dose of dox to achieve the same level of expression, comparing to add dox into culture medium. So, the better choice will be using tet-off system, which can achieve constantly expression without adding dox. For preventing the expression of suicide gene in iPS cells, we only need to add dox during the in-vitro differentiation.
However, combining a special suicide gene apoptin, which will not kill normal cells when constantly expressing, with another suicide gene, it is possible to use tet-on system without constantly intake the dox in vivo. And the time of adding dox will be shorter.
One, two, three: which is the golden generation?
There are three generations of tet-expression systems now [15].
We expect that our suicide gene has low level of leakage expression and a high fold increasement. First, we want to find out the one with lowest leakage expression. It turns out to be tTA+p_tight. However, a low leakage expression is usually in accompany with low maximal expression. Hence, transient transfection proved that the suicide gene is not strong enough to kill all cells. But cells escaped from surveillance might go through rapid genetic rearrangement to acquire invasiveness. Actually we have gotten another type of Tet-off system fused with KRAB transcription factor [16]. It has a higher leakage expression but the fold increasement is the best. Actually, this problem can be circumvented in the process of generating stable cell lines because leakage expression differs from clone to clone and is lower than in transient transfection. However, KRAB does not function in ES cells and this Tet-off is abandoned. We are also interested in alternative Tet system like those controlling the expression of RNA and the else.

Figure 7. Mechanism of a new tet system.
pEF-1α or pCMV
To consistently express the protein tTA of our circuit in a high level, we finally select a human elongation factor 1 alpha ( PEF-1α ) to replace the original promoter pCMV to be our promoter in tet-off system.
Generous & strong
Since Elongation Factor 1α takes part in translation, almost every types of mammalian cells need to express this factor. Hence its promoter, EF-1alpha, is constantly activated no matter what kind of state the cell is undergoing [17]. In our design, the expression of our device is supposed to be stable and persistent during the whole differentiating process, so we choose EF-1alpha promoter to drive tTA transcription. Besides, EF-1alpha promoter is one of the strongest promoters that has been used in constructing mammalian circuit.
No silence in PSCs
EF-1alpha promoter has been proved to be particularly useful in constructing stable cell lines. Compared to another strong promoter, CMV promoter, which has also been widely used in constructing mammalian circuit, EF-1alpha promoter won't undergo transgenic silence in certain cell types, including the iPS cells in our project and hematopoietic stem cells et al [18].
Previous work has showed that the expression level of gene driven by EF-1alpha promoter is more robust in several cell lines, which will provide convenience to construct general circuit used in different cell lines. The data that characterize in one type of cell lines will be reliable in other types.
lentivrus
We decided to use lentivirus vector to carry our circuits, deliver and long-term maintain of our circuit in our chassis genome. We will introduce the mechanism of lentiviral transfection system and the reasons why we choose it.
What is lentivirus
Lentiviral vectors (LV) are viral-based gene delivery systems that can stably deliver genes or RNAi into primary cells or cell lines with up to 100% efficiency. LVs bind to target cells using an envelope protein which allows for release of the LV RNA containing the gene or gene silencing sequence into the cell. The LVs RNA is then converted into DNA using an enzyme called reverse transcriptase by a process called reverse transcription. The DNA pre-integration complex then enters the nucleus and integrates into the target cell's chromosomal DNA. Gene delivery is stable because the target gene or gene silencing sequence is integrated in the chromosome and is copied along with the DNA of the cell every time the cell divides. One of the discriminating features of LVs is their ability to integrate into non-dividing cells, in contrast to other vectors that either don't integrate efficiently into chromosomal DNA (e.g. non-viral, Adenoviral and Adenoviral-Associated vectors) or can only integrate upon cell division (e.g. conventional Retroviral vectors).
Why lentivirus
Because our device is supposed to be expressed stably in a long term and therefore persistently safeguard the cells under any condition, we finally choose lentivirus to be our delivery system into iPSCs, for its high transfection efficiency and the ability to integration into host genome. Though we can never escape the fact that the use of lentivirus could increase the oncogenic risks in cells at the same time, however, considering another fact that almost every inducing/reprogramming factors in iPSCs are oncogenes, maybe the better thing we could do here is to deliver a monitor device which would persistently safeguard the cells in a long term.
To sum up, we choose lentivirus as our delivery system mainly for the following reasons:
-
- Lentivector can integrate into human genome in a multi-copy way.
- Lentivector can stably expressed in wide types of cell lines, without being silenced or irreversible transgenic suppression in ESCs and iPSCs.
- Lentivector has a relatively higher efficiency compared with other integrating virus.
- Lentivector is relatively safer in all integrating virus.
The lentivector we used in our project is the 3rd generation inducible lentivirus pPRIME, whose packaging plasmids are psPAX2 and pMD2G. We packaged lentivirus mix in 293T cell lines.
All parts together!
Up till now, we have introduced all the gene parts. Combining them toghther, our cuicurt finally works like this:

Figure 8. Mechanism of our final curcuit. iPSCs are firstly cultured with Dox. Once we want to turn on our device, Dox will be removed,after which the expression level of endogenous miRNA122 will be the only key. When iPSCs are successfully differentiated into hepatocytes, high level of miRNA122 will bind to the targets and therefore slience suicide mRNA tranlation. However, when tumorigenesis happens in the cell, the level of miRNA122 drop significantly, thus the suicide protein will finally lead to cell apopotosis. Therefore, those tumor cells are picked out from the iPSC mass.
References
[1] Amotz Nechushtan et al, The EMBO Journal,18,(1999)
[2] Zhen Xie et al.,science, Science 333, 1307 (2011)
[3] Qunli Xu et al,molecular cell, 1,3, 1998
[4] ]AG Porter et al,cell death and differentiation,6,2,(1999)
[5] Dattananda S. Chelur et al,PNAS,104, 7,2007
[6] Xuanyong Lu etal, Int. J. Med. Sci.,2,1 ,(2005)
[7] Srinivasa M. Srinivasula et al,, J. Biol. Chem,273,10107,(1997)
[8] N Festjens,Cell death and differentiation,14,400,(2007)
[9] Liming Sun et al,Cell, 148,213,2012
[10] Liming Sun et al,Cell, 148,213,2012
[11] Su-Xia Han et al,14,23,(2008)
[12]http://www.clontech.com/CN/Products/Cell_Biology_and_Epigenetics/RNA_Interference/MicroRNA/Tet-Inducible_MicroRNA?sitex=10022:22372:US
[13] Hui Xu et al,Hepatology,52,4,(2010)
[14] Takanori Takebe et al, Nature,499,481(2013)
[15] http://www.clontech.com/CN/Products/Inducible_Systems/Tetracycline-Inducible_Expression/Tet-On_3G?sitex=10022:22372:US
[16] Jolanta Szulc et al,nature method, 3,2 (2006)
[17] Negrutskii BS, Prog Nucleic Acid Res Mol Biol.,60,47,(1998)
[18]http://www.clontech.com/CN/Products/Inducible_Systems/Tetracycline-Inducible_Expression/Tet-On_3G_EF1-Alpha_Promoter?sitex=10022:22372:US
Sun Yat-Sen University, Guangzhou, China
Address: 135# Xingang Rd.(W.), Haizhu Guangzhou, P.R.China