 "
"
Team:York UK/Notebook.html
From 2013.igem.org
| (105 intermediate revisions not shown) | |||
| Line 15: | Line 15: | ||
<div id="container"> | <div id="container"> | ||
| - | |||
<div id="navlocal"> | <div id="navlocal"> | ||
<ul> | <ul> | ||
<li>Notebook</li> | <li>Notebook</li> | ||
| - | |||
<li><div id="drylab"><a href=''>Dry Lab</a></div></li> | <li><div id="drylab"><a href=''>Dry Lab</a></div></li> | ||
| + | <li><div id="wetlab"><a href=''>Jonas' Lab Book</a></div></li> | ||
| + | <li><div id="labbook"><a href=''>Kobchai's Lab Book</a></div></li> | ||
| + | <li><div id="niklabbook"><a href=''>Nikola's Lab Book</a></div></li> | ||
<li><div id="protocols"><a href=''>Protocols</a></div></li> | <li><div id="protocols"><a href=''>Protocols</a></div></li> | ||
| - | <li><div id=" | + | <li><div id="modelling"><a href=''>Modelling</a></div></li> |
| + | |||
</ul> | </ul> | ||
</div> | </div> | ||
<div id="conlocal"> | <div id="conlocal"> | ||
| - | <div id="wetlabcontent" | + | <div id="wetlabcontent" class="hiddenContent"> |
| - | + | ||
<h2>July 10th</h2> <p>Purification of genomic DNA from <i>Shewanella oneidensis</i> and <i>Salmonella sp.</i></p> | <h2>July 10th</h2> <p>Purification of genomic DNA from <i>Shewanella oneidensis</i> and <i>Salmonella sp.</i></p> | ||
| - | <h2>July 25th</h2> <p>After a long break we finally started working on the actual assembly of parts - the mtr complex. Today Jonas ran a trial PCR on mtrB and mtrC part of the complex. GoTaq polymerase (Promega) was used, this is the polymerase we generally use for trials and colony PCRs as fidelity is not the main priority.</p> | + | <h2>July 25th</h2> <p>After a long break we finally started working on the actual assembly of parts - the mtr complex. Today Jonas ran a trial PCR on mtrB and mtrC part of the complex both of which are around the size of 2kb. GoTaq polymerase (Promega) was used, this is the polymerase we generally use for trials and colony PCRs as fidelity is not the main priority.</p> |
| - | <p><img src="https://static.igem.org/mediawiki/2013/c/cc/Gel-mtrBC.jpg" width="340" height="150" alt="mtrBCtrial" align="left | + | <p><img src="https://static.igem.org/mediawiki/2013/c/cc/Gel-mtrBC.jpg" width="340" height="150" alt="mtrBCtrial" align="left">This is the gel from the PCR. Temperatures ranged 46-71˚C. It appears that the primers are sufficiently specific at even low temperatures. 1% agarose, SYBR-Safe(Invitrogen), <a href="http://www.york-bio.com/QStep4.jpg">QStep4 ladder.</a></p> |
| - | <br clear=all><p><h2>July 26th</h2></p> <p>Trial PCR again, this time for three synthetic DNA fragments - gBlocks (IDT), these fragments are highlighted below on the diagram as well as the overall view of our mtr gene. | + | <br clear=all><p><h2>July 26th</h2></p> <p>Trial PCR again, this time for three small (~500bp) synthetic DNA fragments - gBlocks (IDT), these fragments are highlighted below on the diagram as well as the overall view of our mtr gene. </p><p> <img src="https://static.igem.org/mediawiki/2013/8/81/MtrABC.jpg" width="680" height="51" alt="mtrABC"></p> |
| - | </p></div> | + | <p><img src="https://static.igem.org/mediawiki/2013/4/43/Gel-mtrA.jpg" width="340" height="150" alt="mtrAtrial" align="left" >The gel. Three different temperatures (51-61˚C)- all work. 1% agarose dyed with SYBR-Safe, however, due to poor practice the ladder is indistinguishable.</p> <p>At this point Jonas arrived at the realisation that as long as the primers are specific enough, there is no point in doing PCR trials (silly Jonas). Therefore most of the PCRs done this day forth use a universal 58˚C temperature.</p> |
| - | <div id="drylabcontent"><p> | + | <br clear=all><h2>July 27th</h2><p>This is the first day that we actually got some positive outcome from our cloning. Or so we thought.</p><p>All the fragments of our mtr complex were once again amplified via PCR but this time using Phusion polymerase for high fidelity (Thermo Fisher). Gel confirmed that everything went fine and specifically. Using QIAquick PCR purification kit (Qiagen) we clean up the PCR product, then measure the concentrations using NanoDrop1000, do the required calculations and then proceed to the main reaction:</p><p>In 60 minutes at 50 ˚C we joined all of the 5 fragments using the Gibson Assembly Master Mix provided by NEB. Confirmed by a PCR.</p><p> <img src="https://static.igem.org/mediawiki/2013/5/54/Gel-mtrGibson1.jpg" width="120" height="250" alt="mtrGibson1" align="left" >The PCR was not fully specific but it is unclear whether these fragments formed during Gibson reaction (main fragment joining sites are identical RBS) or because the temperature used was too low (51˚C & 58˚C). 3rd and 4th lanes are negative controls. 1% agarose dyed with SYBR-Safe, <a href="http://www.york-bio.com/QStep4.jpg">QStep4 ladder.</a></p> |
| - | <div id="protocolscontent"><p> | + | <br clear=all><p><h2>Before August 11th</h2></p><p>During this time we tried putting all of our genes inside the pSB1C3 iGEM plasmid but with little luck. Everything seemed to be working fine up until the point when we realised that colony PCRs are not so trustworthy. It appeared clear that most of the colony PCRs came out as false positives after we decided to run the PCR on already purified plasmids. However, some samples were already on their way for sequencing (thinking that we actually got something within our pSB1C3 backbones we sent them). The sequencing results gave us an answer: we planned it all wrong! Graphical summary:</p><p><img src="https://static.igem.org/mediawiki/2013/4/43/Themistake.jpg" width="680" height="307" alt="mistake"></p><p>So all of our parts that were to be put inside the iGEM plasmid only had XbaI and SpeI cutting sites, but we were unaware that they actually had complementary sticky ends and it was nearly impossible to ligate something in between.</p><p>We fixed it with new primers which add the remaining part of the prefix and suffix. However, we lost 2 weeks worth of productive labwork.</p> |
| - | <div id=" | + | <h2>August 12th</h2><p>So after receiving the new primers we continue working. This time we're using Gibson's assembly to directly insert our mtr part into pSB1C3, this allows us to skip any problems that might occur during digestion or ligation.Note that during design of these primers we decided to produce two different mtr parts, one of which would lack the promoter and the first RBS(crR12) which we thought might not be as convenient for other teams.</p> |
| - | + | <p>So today we tried amplifying mtrAB(from the previous Gibson), mtrC(from previous PCR, should have used the genome) and pSB1C3 backbone(the linearised plasmid from iGEM HQ). Unfortunately, the backbone PCR failed.</p> | |
| - | + | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/0/05/Mtrabc_and_psb1c3_fail.jpg" width="177" height="278" align="left" alt="psb1c3fail">We thought that perhaps dimers of the primers formed as the sequences of prefix and suffix are not that different. 1% agarose dyed with SYBR-Safe, <a href="http://tools.lifetechnologies.com/content/sfs/gallery/high/2852.jpg">1kb ladder(Invitrogen)</a>(on the right).</p> <p>However, after playing a bit with DMSO and annealing temperatures we were still unable to amplify it until we changed the template. We took a random iGEM part we found in the fridge and tried the PCR on it and it worked. Conclusion: iGEM HQ should provide more information about their plasmid linearisation and how were the ends modified.</p> | ||
| + | <br clear=all><h2>August 13th</h2><p>2nd and 3rd Gibson Assembly. So we purified the assembly parts from previous PCR and did the calculations to maximise the amount of product. This reaction was also relatively simple as we reduced the number of fragments to 3 and it results in already circular plasmid.</p><p><img src="https://static.igem.org/mediawiki/2013/5/50/Gibson_2nd_3rd_success.jpg" width="114" height="222" align="left" alt="gib2nd3rd"> So while we do the transformations, spending some usual late time in the labs, we also did a PCR using VF2 and VR primers. The gel confirms that reactions were successful. We can see the bands just above the 5kb mark,and as there is only about 130bp difference between these distinct mtr constructs it is impossible to see that on the gel. 1% agarose dyed with SYBR-Safe, <a href="http://tools.lifetechnologies.com/content/sfs/gallery/high/2852.jpg">1kb ladder(Invitrogen)</a>(on the right).</p> | ||
| + | <br clear=all><h2>August 14th</h2><p>So after having some sleep we head back to the lab and do the colony PCR on our transformations. But because Jonas is a very lazy person he used Phusion polymerase (which is as much as 4 times faster than GoTaq) and failed - there is nothing visible on the gel apart from ladder. It is not the first time this happens with Phusion colony PCR, so we try to avoid doing it, it appears though that Phusion requires relatively pure DNA template.</p><p> Nonetheless, we inoculate 2 cultures each of both mtr ORFs only and mtr+promoter constructs and grow them overnight. But it is not all for the day.</p> | ||
| + | <br clear=all><p>Also, today we got the new primers for the rest of the cloning, so we amplified the sequences of gold peptides and gold sensing device adding the rest of prefix and suffix. It all worked but the gel is really really hideous thus it will not appear up in here. We leave the rest for tomorrow.</p> | ||
| + | <h2>August 15th</h2><p>We extract the plasmids from overnight cultures using NucleoSpin Plasmid(Macherey-Nagel) and digest them with ThermoFisher's FastDigest enzymes:</p> | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/1/18/Was_it_really_mtr.jpg" width="346" height="171" align="left" alt="wasitmtr"> Every triplet is: 1st the control, 2nd PstI digest and 3rd the EcoRI & SpeI digest. It is obvious that there is nothing near the size of 5kb visible. However, the 1kb fragment that is visible in double digest shows us that it is actually the template we used for pSB1C3 backbone amplification. 1% agarose dyed with SYBR-Safe, <a href="http://tools.lifetechnologies.com/content/sfs/gallery/high/2852.jpg">1kb ladder(Invitrogen)</a>(on the right).</p><p>So the solution is simple: we digest the Gibson reactions we had with DpnI(Promega) thus removing the background and then repeat the transformations.</p> | ||
| + | <br clear=all><p>At the same time we continued working with other sequences. Amplified gBlocks (peptides,the gold device) were purified with QIAquick kit, digested with EcoRI & PstI and then purified again using the same kit - this step frees our samples of small 2-6bp long fragments making the ligation easier. We also digest the linearised plasmid from iGEM HQ with the same enzymes and purify it as well. Even though this leads us to a very low concentration of pSB1C3, it is still enough for transformations as long as there are no problems with ligations. So we then make the reactions with T4 Ligase(Thermo Fisher) and proceed with the transformations.</p> | ||
| + | <h2>August 16th</h2><p>The day of GoTaq colony PCRs. So basically Gintare marched to the lab to do PCR of 44 colonies (and we are all aware of how annoying it is). Results were pretty straightforward with the peptides and gold sensing device parts. On the left gel below first four are <a href="http://parts.igem.org/wiki/index.php?title=Part:BBa_K1127000">BBa_K1127000</a>, then <a href="http://parts.igem.org/wiki/index.php?title=Part:BBa_K1127001">BBa_K1127001</a> and <a href="http://parts.igem.org/wiki/index.php?title=Part:BBa_K1127003">BBa_K1127003</a>. On the second gel: <a href="http://parts.igem.org/wiki/index.php?title=Part:BBa_K1127002">BBa_K1127002</a>, followed by two constituents (which we join together later) of <a href="http://parts.igem.org/wiki/index.php?title=Part:BBa_K1127008">BBa_K1127008</a> and then <a href="http://parts.igem.org/wiki/index.php?title=Part:BBa_K1127010">BBa_K1127010</a> which was not submitted. | ||
| + | Two colonies of each were cultivated overnight.</p> <p><img src="https://static.igem.org/mediawiki/2013/8/86/Colony_pcr_yg1yg2yg3.jpg" width="350" height="204" align="left" alt="yg1yg2yg3"><img src="https://static.igem.org/mediawiki/2013/5/5c/Colony_pcr_yg5yg71yg7byg8.jpg" width="352" height="208" align="middle" alt="yg5yg7abyg8"></p> | ||
| + | <br clear=all><p>However, the results for mtr complexes were weird. So we picked a few cultures and grew them overnight for further analysis.</p> <p> <img src="https://static.igem.org/mediawiki/2013/b/b4/Colony_pcr_mtr_only.jpg" width="295" height="218" align="left" alt="cpcrmtronly"><img src="https://static.igem.org/mediawiki/2013/9/9b/Colony_pcr_mtr_luxI.jpg" width="312" height="218" align="middle" alt="cpcrluximtr"></p> | ||
| + | <br clear=all><p>All gels were 1% agarose dyed with SYBR-Safe, <a href="http://tools.lifetechnologies.com/content/sfs/gallery/high/2852.jpg">1kb ladder(Invitrogen)</a>(on the right). We also noticed that something was not right with our gel viewing box as the images turned out to be very dark even at max exposure.</p> | ||
| + | <h2>August 17th</h2><p>Another busy day, so firstly about mtr complex. We purified the plasmids from yesterdays colonies using our usual NucleoSpin Plasmid kit and then digest them with FastDigest enzymes:</p><p> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/4/4a/Sdm_1st.jpg" width="370" height="247" align="left" alt="1stsdm"></p> <p>Triplet consists of: 1. Control 2. PstI digest 3. EcoRI & SpeI digest. First three triplets are of the mtr ORFs only construct and the last one (a lot of it!) is the one with promoter. Even though the gel is faint we can see the most important thing: double digests separate the pSB1C3(2kb) backbone and our part (5.3kb). The PstI digest also confirms the prohibited PstI cutting site within mtrC sequence (present in the genome). 1% agarose dyed with SYBR-Safe, <a href="http://tools.lifetechnologies.com/content/sfs/gallery/high/2852.jpg">1kb ladder(Invitrogen)</a>(on the right).</p> | ||
| + | <br clear=all><p>Using the primers we designed earlier we do the Site-Directed Mutagenesis on each of the mtr constructs, which should remove the PstI cutting site from where it should not be present but still retaining the aminoacid sequence. We also finish it up with DpnI digestion to remove any background and then transform the cells.</p> | ||
| + | <br clear=all><p>In the meantime, we did similar to mtr digestions on the peptide parts we cloned earlier. Again on NucleoSpin purified plasmids with FastDigest enzymes.</p><p> <img src="https://static.igem.org/mediawiki/2013/9/91/Peptides_et_br.jpg" width="281" height="192" align="left" alt="peptideetbr"><img src="https://static.igem.org/mediawiki/2013/c/ca/Testing_the_presence_of_peptides.jpg" width="336" height="192" align="middle" alt="peptidesybr"></p><br clear=all><p>Each plasmid was ran on the gel in triplets: 1. uncut control 2. XbaI & SpeI digest 3. EcoRI & PstI digest. Both gels show the same peptides mentioned earlier from different colonies picked yesterday. The gel shows us that the peptides are actually there within the pSB1C3 with working prefixes and suffixes (there was only a slight nuisance with the 3rd peptide). 1% agarose gels dyed with either ethidium bromide(left) or SYBR-Safe(right), <a href="http://www.york-bio.com/QStep4.jpg">QStep4 ladder.</a></p> | ||
| + | <h2>August 18th</h2><p>Today we do colony PCR (yet again!) on our SDM'd plasmid. A clever way we thought of to check if PstI cutting site is still there was to amplify the mtrC part of the colony and digest it immediately with PstI:</p><p><img src="https://static.igem.org/mediawiki/2013/f/fa/Sdm_improved_pcr.jpg" width="246" height="112" align="left" alt="sdmpcr"> Although it was not so cleverly thought to the end: Jonas forgot the control for mtrC before the SDM. Basically, the first pair is the control on the SDM reagent from yesterday, the rest of them are our mtr constructs. First member of the pair is undigested product and the other one is PstI digested product. We can see the the bands are not at the equal sizes, however, that's an optical illusion caused by different volumes added to the lanes (yes, we checked under the manual UV lamp). 1% agarose dyed with SYBR-Safe(right), <a href="http://www.york-bio.com/QStep4.jpg">QStep4 ladder</a>(although irrelevant).</p> | ||
| + | <br clear=all><p> We also tried joining the two mentioned parts to produce <a href="http://parts.igem.org/wiki/index.php?title=Part:BBa_K1127008">BBa_K1127008</a> using <a href="http://www.jbioleng.org/content/5/1/17">Amplified Insert Assembly</a>.</p> | ||
| + | <h2>August 19th</h2><p>And it did not work for us.</p><p><img src="https://static.igem.org/mediawiki/2013/e/e2/Aiafail.jpg" width="419" height="194" align="left" alt="aiafail">This is the colony PCR with VF2&VR primers and all the bands are clearly lower than the expected 900bp size, it actually fits the size of our recipient plasmid. 1% agarose dyed with SYBR-Safe, <a href="http://www.bioline.com/Images/HyperLadder/HyperLadder_1kb_550.jpg">Hyper ladder 1kb(Bioline).</a></p><br clear=all><p>We spent the remaining time designing primers for the sequencing of whole mtr complex.</p> | ||
| + | <h2>August 22nd</h2><p>After trying a few other techniques and failures of joining our two fragments together (note that they are now in biobrick format) we decided to test something new. Basically we joined the parts together before putting them inside the plasmid:</p><p><img src="https://static.igem.org/mediawiki/2013/3/3d/Yorkassembly.jpg" width="510" height="348" align="left" alt="yorkassembly"></p><br clear=all> | ||
| + | <h2>August 23rd</h2><p>We select two random colonies from yesterday transformations and grow them overnight.</p><p>Also, we receive sequencing primers for mtr and send the samples to <a href="http://www.lifesciences.sourcebioscience.com/">Source BioScience.</a></p> | ||
| + | <h2>August 24th</h2><p>Purified plasmids from cultures are cut to confirm that we have the gold sensing particle ready.</p><p><img src="https://static.igem.org/mediawiki/2013/c/c4/7abdigest.jpg" width="270" height="186" align="left" alt="7abdigest">Left and right of the marker are two plasmids: first the uncut control, XbaI digest, SpeI digest, EcoRI&PstI double digest followed by SanDI digest. Double digest reveals the ~750bp long fragment which proves our method successful. 1% agarose dyed with SYBR-Safe, <a href="http://www.bioline.com/Images/HyperLadder/HyperLadder_1kb_550.jpg">Hyper ladder 1kb(Bioline).</a></p> | ||
| + | <br clear=all> | ||
| + | <h2>August 27th</h2><p>We receive our first sequencing results of mtr complexes. Unfortunately, we found a single base pair substitution at the very start of mtrA (changing the aminoacid lysine to glutamic acid) and a nonsense mutation in mtrC. However, the mtr construct with promoter contained an additional nonsense mutation within mtrA, this is the point where we decided to give up on this one and try and fix the other, more sensible biobrick.</p><p>So we ignore the single base pair substitution (as it is not part of the protein motive compared with other mtrA analogs) and order new set of primers for the Site-Directed Mutagenesis of mtrC.</p> | ||
| + | <h2>August 30th</h2><p>We receive the primers for mtr SDM from IDT and do it immediately the same way we did it last time.</p><p>Samples for sequencing of the peptides and gold sensing constructs were sent today.</p> | ||
| + | <h2>September 4th</h2><p>We do the β-galactosidase assay on the gold sensing device for which the results can be found elsewhere.</p><p>The mtr part was sent for sequecing again to see if the SDM was successful.</p> | ||
| + | <h2>September 10th</h2><p>Finally, DNA of the parts for submission is prepared and is finally sent to iGEM HQ on September 16th.</p> | ||
| + | |||
| + | |||
| + | |||
| + | </div> | ||
| + | <div id="drylabcontent" class="hiddenContent"> | ||
| + | |||
| + | <h2>February 21st</h2> | ||
| + | <p>Today the iGEM team divided to discuss about the two main ideas, chosen from over 100, after meeting with potential supervisors. </p> | ||
| + | <p>The first idea is making a mouthwash which contains harmless, live bacteria which normally reside in the mouth. These bacteria will be genetically engineered to secrete pleasant odours (e.g. menthol, rose, banana - parts for which have already been submitted to the iGEM registry). The scope is to have the bacteria colonise the mouth; to prevent the smell being secreted at all times, their production could be controlled by controlling gene expression, depending on external signals (such as concentration of oxygen, glucose or stress). </p> | ||
| + | <p>Becca, Evaldas, Andy, Jonas, Andra, Hannah and Caroline looked into the idea in more depth. One of the major concerns is that safety issues would prevent the mouthwash from being regarded as a product which can be used, since it involves introducing live, genetically modified bacteria into humans. Also, the idea is not conceptually very challenging.</p> | ||
| + | <p>The second idea is to create an improved microbial fuel cell, a type of battery that uses live bacteria for current production. The bacteria releases H ions and electrons into the environment, when encapsulated in an anode chamber. These secreted particles contribute to energy formation, when electrons pass from the anode to the cathode of the battery. We would like to genetically engineer <i>E.coli</i> to secrete more electrons, by introducing the secretion system present in a different organism, <i>Shewanella oneidensis.</i> This has been attempted in iGEM before, but never achieved. In addition, we would try to deposit gold nanoparticles on the anode of the battery, to increase its conductivity. </p> | ||
| + | <p>Ivan, Kobchai, Nick, Ric, Gintare, Rudolfs and Alex looked into this idea. The main challenges are that, although nanoparticles have been created before, we are unsure which genes we would use for this process. More reading through literature is needed to identify them, before carrying on.</p> | ||
| + | <h2>February 27th</h2> | ||
| + | <p>Each subteam presented their latest findings for the two ideas. Kobchai went to discuss with people from the engineering department, who gave useful insight into how nanoparticles are made in vivo and which organisms and gene could be used. We found out that <i>Salmonella enterica</i> has a good gold-detection system, which we could use to control gene expression in the presence of gold ions. We have also identified a few papers which describe how bacteria secrete nanoparticles. We decided this would be a good starting point to carry on with the gold idea.</p> | ||
| + | <p>We have given ourselves two weeks to divide work into subgroups and do reading on several issues; 1st year students will be grouped with older students, to ensure they receive support in interpreting data and understanding the articles. To do list: | ||
| + | <br> ● what genes would be involved in electron secretion? | ||
| + | <br> ● what method of nanoparticle formation would we use? | ||
| + | <br> ● how would we control gene expression? | ||
| + | <br> ● are there any parts in the iGEM Registry we could use?</p> | ||
| + | <h2>March 13th</h2> | ||
| + | <p>Ivan gave a comprehensive presentation on our findings so far, in front of potential supervisors. We have established that the main objective of our project is to improve existing microbial fuel cell technology and make fuel cells more cost effective. The three aims which lead to this are: | ||
| + | <br> 1) Expressing the MTR protein complex from Shewanella on the surface of <i>E.coli</i> for electron transport; | ||
| + | <br> 2) Creating a MFC of approx 3cm diameter and engineer <i>E.coli</i> to deposit gold nanoparticles on the anode; | ||
| + | <br> 3) Use a quorum sensing system to control gene expression of genes involved in gold deposition.</p> | ||
| + | <p>Ideas which emerged during the meeting concerned the following: | ||
| + | <br> ● would the electron transporter introduced affect bacterial metabolism? | ||
| + | <br> ● would the bacteria form biofilms on the anode? Could this be avoided by expressing pilli? | ||
| + | <br> ● what is the gold concetration needed and where would we take it from? Possible solution: pharmaceutical waste rich in gold ions, which would also allow the safe removal of toxic ions from the environment | ||
| + | <br> ● other metals have been discussed, as alternatives to gold, such as copper and silver. However, there are already genes which are involved in copper metabolism in bacteria, so their effects might interfere with our system. | ||
| + | <br> ● two bacterial strains could be used, living together inside the MFC, one for the electron secretion and one for the quorum sensing</p> | ||
| + | <h2>May 1st</h2> | ||
| + | <p>After the spring holiday, during which the team has worked from home on iGEM, we had a n important meeting with our supervisors.</p> | ||
| + | <p>We are considering using a peptide which can bind elemental gold, such as MIDAS, to create nanoparticles. We have discussed how to ensure the nanoparticles are deposited on the anode. There were several variants proposed: | ||
| + | <br> ● fuse the peptide to secreted protein and let the complex settle on the anode (by gravity or magnetism) | ||
| + | <br> ● fuse the peptide to surface proteins (such as OpmA) and then as above | ||
| + | <br> ● turn the whole chamber into a graphyte anode, to increase the chance of bacteria being trapped in the porous material and release nanoparticles there | ||
| + | <br> ● lyse bacteria once they have captured gold ions internally, to release the peptide, and then repopolate the MFC with the second bacterial strain, which secretes the MTR complex | ||
| + | <br> ● aqua regia was proposed as a source of gold ions for the initial stages of the project, as an alternative to pharmaceutical waste; the composition of the latter may vary between experiemtns and will prevent us from making controlled, accurate measurements between replicate experiments</p> | ||
| + | <h2>May 15th</h2> | ||
| + | <p>During today's meeting we discussed more the details of the peptides we want to create. We have looked into several secretion tags which we would like to fuse to the peptide. We have narrowed it down to two, for now. HlyA can transport small peptides outside bacteria (though our initial in silico simulations show the peptidic construct would not be very stable). We might need a special <i>E.coli </i>strain if we work with it. pelB is good for small peptides, easy to use and we have an available vector for it. Another option is to have a poly-peptide, made with the A3, nanoparticle-producing peptide, rather than MIDAS. This poly-A3 would be secreted and cleaved outside of the bacteria; this method would give more stability to the system.</p> | ||
| + | <p><b>Need to look more into:</b> | ||
| + | <br> ● Physiology of bacteria in the MFC | ||
| + | <br> ● What we will compare (e.g. two MFC, one improved and one normal) | ||
| + | <br> ● We need to design a good, simple end-assay, which will allow us to test the effect of our peptide on electricity production even if the rest of the system does not work/is not ready yet (e.g., he suggests putting the peptide in <i>Shewanella</i>, if we're not ready closer to the finish, and show it produces NP in <i>Shewanella</i>).</p> | ||
| + | <h2>June 5th</h2> | ||
| + | <p>After exams, we divided into four groups and started doing extensive reading on genes, to start preparing the gene sequences we need to order. Ric, Hannah, Becca and Andra are looking into the gold sensing system existing in <i>Salmonella</i>, GolTS. Alex, Evaldas and Jonas are looking into peptide design and secretion. Ivan, Nick and Andy are looking into modelling and identifying genes in the iGEM Registry. Kobchai, Caroline and Rudolfs are looking into the quorum sensing system and system regulation as a whole.</p> | ||
| + | <p>Notable things so far: some of the sequences for the pGolTS system submitted by other teams to the Registry seem to be wrong; we need to look more into it during the following week.</p> | ||
| + | <p>We might use some of the parts from team Edinburgh, which were not submitted last year, but which they can send to us, for the mtr complex. </p> | ||
| + | <h2>June 12th</h2> | ||
| + | <p>Evaldas, Kobchai and Andra gave a presentation on what has been done so far. Andy talked about the modelling he worked on, using the kinetic values obtained from scientific papers. . We need to look more into the details of the MFC we are planning on making, in particular what buffers we are planning on keeping the bacteria in, whether we will grow the bacteria in an aerobic or anaerobic environment and what bacterial strains to choose.</p> | ||
| + | <h2>June 19th</h2> | ||
| + | <p>The team spent the week designing some of the constructs, but also looking into the details of the MFC construction. We came together to make some decisions regarding the latter. We have established that, to allow the bacteria to create biofilms, it would be better to grow them in an anaerobic environment in the final MFC. However, for the initial experiments, we will use an aerobic environment. The two bacterial strains we will use are K12 and W3110, as they have been used with MFC before, are well characterised and easily available. </p> | ||
| + | <p>We also met Prof. Smith, to discuss about methods of isolating the peptide once it is produced, for part characterisation. We chose to put a flag tag on the peptide, to be identified with antibodies by western blot.</p> | ||
| + | <h2>June 26th</h2> | ||
| + | <p>During our supervisory meeting we talked about which vectors are best to be used for our constructs, and which constructs should be put on what vectors. We have discussed the advantages of pET vs PUC vectors and other possibilities proposed. We decided to go with the 3 vectors of the pET duet, which have different antibiotic resistance and have compatible origins of replication. We would use one for the gold recognition, one for mtr and one for the quorum sensing. </p> | ||
| + | <p>We also showed the supervisors our defined sequences, hoping we will soon order the sequences. We presented the techniques we are planning to use for every part, to characterise it (beta galactosidase assays). Also, the methods of assembly were presented to the supervisors (amplified insert assembly, which they were unfamiliar with, and the formerly proposed Gibson assembly). We received positive feedback on our choices.</p> | ||
| + | <p>An issue was raised regarding the way in which we would isolate peptides from cells, to assess their presence (since they are too small to be run on a gel). We chose to perfor, pull down assays. Defining sequences. To be expanded. </p> | ||
| + | <h2>July 2nd</h2> | ||
| + | <p>We had a small discussion with Dr. Thomas, about ordering the sequences. We went over all the designed sequences, verified restriction sites and are now preparing to submit the online order for the sequences. We have received the ordered consumables and are now ready to start working in the lab. Our meetings from now on will be administrative meetings and troubleshooting meetings. </p> | ||
| + | <h2>July 16th</h2> | ||
| + | <p>Internal meeting to redistribute the work within the team</p> | ||
| + | <h2>July 24th</h2> | ||
| + | <p>Final meeting with Dr. Chong before he leaves for two weeks. Catch up with supervisors on how lab work is going.</p> | ||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | </div> | ||
| + | <div id="protocolscontent" class="hiddenContent"> | ||
| + | |||
| + | |||
| + | <h2>Rapid Assembly of Biobricks (RAB)</h2><p>We found this easy way of joining two or more biobricks together:</p> | ||
| + | <p>1. Amplify the parts using VF2&VR primers (although specific primers make it easier). | ||
| + | <br>2. Digest one product with SpeI and the other one with XbaI. | ||
| + | <br>3. We would usually gel extract the fragments we need, which makes the next step easier. | ||
| + | <br>4. Ligate the two fragments together. | ||
| + | <br>5. Now amplify with PCR again the whole fragment that should have formed. | ||
| + | <br>6. You can continue joining more parts together by repeating the above steps. | ||
| + | <br>7. Finally, insert your new part into the iGEM plasmid using the standard procedure. | ||
| + | <br>8. Transform.<p> | ||
| + | <p>Now many would argue that this continuous PCR would result in lots of mutations. However, polymerases with already high fidelity are being continuously improved to the point where their mutation rate is hard to detect statistically. Also, we sequence the parts anyway, so there is high potential in this assembly. We successfully joined our gold sensing device using this protocol. If you feel confused about the procedure, perhaps this picture will help to clarify the mechanism:</p> | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/3/3d/Yorkassembly.jpg" width="510" height="348" align="left" alt="RAB"></p> | ||
| + | <br clear=all> | ||
| + | |||
| + | <h2>Beta galactosidase assay for gold induction</h2> | ||
| + | <p><b>Growth:</b> | ||
| + | <br>1. Transfer one colony to LB with antibiotics and grow overnight at +37 °C with shaking. | ||
| + | <br>2. Next morning transfer bacteria to fresh LB with antibiotics up to OD600=0.1 and continue growing until exponential stage (OD600=0.6-0.8). | ||
| + | <br>3. Induce with required concentration of AuCl4 and continue growing at 30 °C or 37 °C depending on the experiment. | ||
| + | <br>4. Samples for measurements can be taken at time points or after overnight growth.</p> | ||
| + | <p><b>Measurements:</b> | ||
| + | <br>1. Take 200 µl of cell suspension and measure OD630 (due to machine that we used instead of OD600) in a 96 well plate. | ||
| + | <br>2. Take 100 µl of cell suspension for beta gal assay and mix with 100ul Z-buffer. | ||
| + | <br>3. Add 10 µl 0.1% SDS and 20 µl chloroform and incubate at 30 °C for 5 min to disrupt the cells. | ||
| + | <br>4. Add 50 µl 5mM PNPG and incubate at 30 °C for 30 min (incubation times can be extended up to 1h if the colour is not appearing, however the same incubation time should be applied for all of the related samples). | ||
| + | <br>5. To stop the reaction and increase the intensity of yellow colour add 100 µl of 1 M Na2CO3 and centrifuge and 13k rpm for 5 min. | ||
| + | <br>6. Measure the absorbance at 405 nm. | ||
| + | <br>7. The relative response (∅) was calculated according to the formula: | ||
| + | <br><img src="https://static.igem.org/mediawiki/2013/1/1e/Theformula.jpg" width="216" height="39" align="middle" alt="theformula"> | ||
| + | </p><br> | ||
| + | <p><b>Reagents:</b></p> | ||
| + | <p>Z-buffer stock solution: | ||
| + | <br>4.27 g Na2HPO4 | ||
| + | <br>2.75 g NaH2PO x 4H2O | ||
| + | <br>0.375 g KCl | ||
| + | <br>0.125 g MgSO4 x 7H2O | ||
| + | <br>Adjust to pH 7.0. | ||
| + | <br>Bring to 500 ml with dH2O. Do not autoclave. Store at 4°C.</p> | ||
| + | <p>For complete Z-buffer - Prior to daily use mix: | ||
| + | <br>50 ml Z-buffer | ||
| + | <br>0.14 ml ß-mercaptoethanol</p> | ||
| + | <p>1 M Na2CO3 (store at 4°C) | ||
| + | <br>5.3 g Na2CO3 | ||
| + | <br>50 ml dH2O</p> | ||
| + | |||
| + | |||
| + | <h2>Bacterial growth media recipes:</h2> | ||
| + | <p><b>Luria-Bertani media</b></p> | ||
| + | <p>10g Tryptone | ||
| + | <br>5g Yeast Extract | ||
| + | <br>10g NaCl | ||
| + | <br>(15g Agar for solid media) | ||
| + | <br>dH2O to 1000ml. Autoclave.</p> | ||
| + | <p><b>M9 minimal media</b></p> | ||
| + | <p>6g NaHPO4 | ||
| + | <br>3g KH2PO4 | ||
| + | <br>0.5g NaCl | ||
| + | <br>Mili-Q water to 975ml. Autoclave.</p><p> | ||
| + | <br>Under sterile conditions: | ||
| + | <br>1ml 1000x vitamins | ||
| + | <br>1ml 1000x trace metals | ||
| + | <br>2ml 1M MgSO4 (autoclaved) | ||
| + | <br>0.2ml 1M CaCl2 (autoclaved) | ||
| + | <br>1ml 1000x antibiotic | ||
| + | <br>20ml 20% glucose (filtered via 0.2 micron filter)</p> | ||
| + | |||
| + | |||
| + | <h2>Preparation of chemically competent cells</h2> | ||
| + | <p><b>Solutions:</b></p> | ||
| + | <p>TFB1: | ||
| + | <br>30mM Potassium Acetate | ||
| + | <br>10mM CaCl2 | ||
| + | <br>50mM MnCl2 | ||
| + | <br>100mM RbCl | ||
| + | <br>15% glycerol | ||
| + | <br>Adjust pH to 5.8. Filter sterilise (0.2µm). Store at room temperature.</p> | ||
| + | <p>TFB2: | ||
| + | <br>10mM MOPS | ||
| + | <br>75mM CaCl2 | ||
| + | <br>10mM RbCl | ||
| + | <br>15% glycerol | ||
| + | <br>Adjust pH to 6.5. Filter sterilise (0.2µm). Store at room temperature.</p> | ||
| + | <p><b>Notes:</b></p> | ||
| + | <p>Warm clothing is recommended for day 3. | ||
| + | <br>Prechill all equipment to be used in the cold room. | ||
| + | <br>When working from day 3 (step 4) keep the cells cold at all times and perform re-suspensions in the cold room. | ||
| + | <br>Make sure to read the protocol thoroughly before starting.</p> | ||
| + | <p><b>Method:</b></p> | ||
| + | <p>1. Streak a non selective LB plate with a dilution of the previous batch of Competent Cells, incubate at 37°C overnight. | ||
| + | <br>2. The next day, inoculate a single colony from the plate in 2.5ml of LB broth. Incubate at 37°C overnight with shaking at approximately 225rpm. | ||
| + | <br>3. The next day, use the entire overnight culture to inoculate 250ml of LB medium containing 20mM MgSO4 (a 1/100 dilution of 2mM stock). Take 1ml of the culture and keep in a cuvette, as blank for OD measurement. Grow the cells in an autoclaved 1L flask until the OD600 reaches 0.4-0.6 (check after 2 hours then keep testing until within the range). A 1L flask is necessary for proper aeration and growth. | ||
| + | <br>4. Pellet the cells by centrifugation at 4500g for 5 minutes at 4°C. For a 250ml culture use two 250ml centrifuge bottles in a large rotor (eg. Beckman), or if not available use 50ml tubes in a bench top centrifuge at 3500rpm for 15 minutes at 4°C. Tubes need to be prechilled! | ||
| + | <br>5. Re-suspend the cell pellets in 0.4 volume (based on the original culture volume) of ice cold TFB1, using vortex, in the cold room. For a 250ml subculture, use 100ml of TFB1 (50ml/bottle). Combine the re-suspended cells in one bottle. For the remaining steps, keep the cells on ice and chill all pipettes, tubes and flasks. | ||
| + | <br>6. Incubate the re-suspended cells on ice for 5 minutes at 4°C. | ||
| + | <br>7. Pellet the cells by centrifugation as per step 4. When transporting cells to centrifuge, keep them on ice. | ||
| + | <br>8. Re-suspend the cells in 1/25 of the original culture volume of ice cold TFB2, using vortex, in the cold room. For a 250ml subculture use 10ml of TFB2. | ||
| + | <br>9. Incubate the cells on ice for 60 minutes and then aliquot 100µl/ Eppendorf tube for storage at -80°C. | ||
| + | <br>10. Quick freeze the tubes in a dry ice/isopropanol bath (or liquid nitrogen). Cells should be stable for between 3 months to 1 year.</p> | ||
| + | <p><b>Transformation:</b></p> | ||
| + | <p>1. Thaw the competent cells on ice. Use 50µl per transformation. | ||
| + | <br>2. Gently mix the cells with up to 5µl of DNA without pipetting up and down. | ||
| + | <br>3. Incubate on ice for 20 minutes. | ||
| + | <br>4. Heat shock the cells at 42°C for 45s. | ||
| + | <br>5. Place the cells on ice for 3-5 minutes. | ||
| + | <br>6. Add 200-950µl of room temperature LB or SOC media. | ||
| + | <br>7. Incubate the cells at 37°C whilst shaking them at 225rpm for 1hour (2 hours if plasmid contains chloramphenicol resistance). | ||
| + | <br>8. Spread up to 200µl on the selection plates and incubate at 37°C overnight.</p> | ||
| + | |||
| + | |||
| + | <h2>Colony PCR</h2> | ||
| + | <p>1. Combine the master mix for PCR on ice (20µl reactions are sufficient). | ||
| + | <br>2. Dip the pipet tip to the selected colony and now either: | ||
| + | <br>a) Pipet up and down directly into PCR mix aliquot and store the tip inside eppie in the fridge, or | ||
| + | <br>b) Pipet the bacteria into 10µl of H2O and pipet 1µl into the reaction, storing the rest in the fridge | ||
| + | <br>3.Continue the PCR as normal and run the gel, you can then grow the selected cultures from the bacteria you saved in the fridge.</p> | ||
| + | |||
| + | <h2>Site-Directed Mutagenesis</h2> | ||
| + | <p>1. Design the primers with mutation that meet end to end (there is software for that) and have them synthesized 5' phosphorylated (SDS-PAGE purification recommended). | ||
| + | <br>2. Perform a PCR as usual with high fidelity polymerase. | ||
| + | <br>3. Recircularise the amplified plasmid with DNA ligase. | ||
| + | <br>4. Digest with DpnI. This is optional if you use little amounts of template (as low as 10pg) and are going to send 3 or more samples for sequencing. | ||
| + | <br>5. Transform.</p> | ||
| + | |||
| + | </div> | ||
| + | <div id="modellingcontent" class="hiddenContent"> | ||
| + | <p><h2>Models for the gold sensing device <a href="http://parts.igem.org/Part:BBa_K1127008">(BBa_K1127008)</a></h2></p> | ||
| + | <p>Solutions to this problem are relatively simple. The law of mass action which is widely used in biochemistry, physiology and pharmacology is one of the straightforward approaches. However, it requires a massive amount of data at different points in time. This is very tricky to maintain accuracy and to spend less resource. Fitting pre-existing models in our data came to us as a plausible solution. So we measured activities of the device when it reached steady states. Data were collected and fit into the nonlinear models for comparison.</p> | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/e/eb/Formulaone.jpg" width="50%" align="middle" alt="formulaone"></p><br clear=all> | ||
| + | <p>We computed information Akaike Information Criterion (AIC) to determine quality of the models. Regardless of values of the goodness of fit, Logistic is the best model among the three. So model parameters of the Logistic are estimated and shown here.</p> | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/b/b3/Tablemodel.jpg" width="50%" align="middle" alt="tablemodel"></p><br clear=all> | ||
| + | <p>The model is simulated and compared with the experimental data. As seen in the Figure 1, Logistic is well fit and could be a good reflection of reality.</p> | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/9/94/Graphone.jpg" width="50%" align="middle" alt="graphone"></p><br clear=all> | ||
| + | <p>The model is extended to include diffusion of the gold. 'e' is the diffusion constant. In the other word, it is the permeability of the bacterial envelope for the ion Au(III). We solved the differential equations and produced simulations to predict behaviour of the system in various conditions. </p> | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/e/e4/Formulatwo.jpg" width="50%" align="middle" alt="formulatwo"></p><br clear=all> | ||
| + | <p>The model predicts multiple steady. As seen in the Figure 2, the system could reach higher steady states with increasing gold concentration.</p> | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/8/8c/Graphtwo.jpg" width="50%" align="middle" alt="graphtwo"></p><br clear=all> | ||
| + | <p>In Figure 3, the system is fairly robust. It could approach the same steady state despite of differences in the diffusion constants.</p> | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/b/b6/Graphthree.jpg" width="50%" align="middle" alt="graphthree"></p><br clear=all> | ||
| + | <p>For the sake of curiosity, we include expression of the synthetic peptide for gold bio-mineralization. Extension of the model is shown below.</p> | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/4/48/Formulathree.jpg" width="50%" align="middle" alt="formulathree"></p><br clear=all> | ||
| + | <p>Literature about this peptide is scarce, so we make following assumptions: | ||
| + | <br>1. <img src="https://static.igem.org/mediawiki/2013/9/99/Smallformulaone.jpg" alt="smallformulaone">is the change in expression of the peptide; | ||
| + | <br>2. Gold bio-mineralization is a linear process with the constant rate 'f'; | ||
| + | <br>3. Secretion of the peptide is in the form of standard Hill equation <img src="https://static.igem.org/mediawiki/2013/3/34/Smallformulatwo.jpg" alt="smallformulatwo"> with Hill coefficient=1; | ||
| + | <br>4. The peptide is degraded through a linear process i[peptide]<sub>out</sub>.</p> | ||
| + | <p>The model is simulated and shown in Figure 4. It suggests that the system could undergo the decline phase. The rates of reduction could vary depending on the rates of bio-mineralization.</p> | ||
| + | <p><img src="https://static.igem.org/mediawiki/2013/f/f7/Graphfour.jpg" width="50%" align="middle" alt="graphfour"></p><br clear=all> | ||
| + | <p>Our models can't produce precise numerical solutions of the real world problems. However, they have capabilities to describe structures and to generate broad outcomes to what might happen in certain conditions. This qualitative approach is actually advantageous as it provides guidance for experimental design and data analysis without overloading us with unnecessary information.</p> | ||
| + | |||
| + | |||
| + | <p><h2>R Source code</h2></p> | ||
| + | <pre><code>#Model fitting | ||
| + | library(minpack.lm) | ||
| + | library(bbmle) | ||
| + | library(plyr) | ||
| + | library(ggplot2) | ||
| + | |||
| + | #Observed data between 0uM and 10uM | ||
| + | x<-c(0,0,0,0.5,0.5,0.5,1,1,1,2.5,2.5,2.5,5,5,5,7.5,7.5,7.5,10,10,10) | ||
| + | y<-c(0.002857143,0.00152381,0.00212963,0.011598174,0.009054726,0.011746032,0.021904762,0.00854902,0.016464646,0.038600823, | ||
| + | 0.043166667,0.043508772,0.045142857,0.045367965,0.035365854,0.043474178,0.044122807,0.04971831,0.03822807,0.045063291, | ||
| + | 0.043288889) | ||
| + | #0.043508772<=0.053508772, 0.04971831<=0.05971831, 0.03822807<=0.033? | ||
| + | df<-data.frame(x, y) | ||
| + | |||
| + | #HILL FUNCTION | ||
| + | Hill<-nlsLM(y~((a+b*x^d)/(c+x^d)), start=list(a=0.002, b=0.044, c=1, d=2), trace=TRUE) | ||
| + | |||
| + | #LOGISYIC FUNCTION | ||
| + | Logt<-nlsLM(y~(a+b/(1+exp(4*c*(d-x)/b+2))), start=list(a=0.001, b=0.044, c=0.025, d=0.1), trace=TRUE) | ||
| + | |||
| + | #GOMPERTZ FUNCTION | ||
| + | Gomp<-nlsLM(y~(a+b*exp(-exp(c*2.71828*(d-x)/b+1))), start=list(a=0.001, b=0.044, c=0.025, d=0.1), trace=TRUE) | ||
| + | |||
| + | |||
| + | AICtab(Hill, Logt, Gomp) | ||
| + | # dAIC df | ||
| + | #Logt 0.0 5 | ||
| + | #Gomp 1.8 5 | ||
| + | #Hill 3.6 5 | ||
| + | |||
| + | |||
| + | summary(Logt) | ||
| + | #Formula: y ~ (a + b/(1 + exp(4 * c * (d - x)/b + 2))) | ||
| + | |||
| + | #Parameters: | ||
| + | # Estimate Std. Error t value Pr(>|t|) | ||
| + | #a 0.0001397 0.0048826 0.029 0.977507 | ||
| + | #b 0.0434417 0.0053800 8.075 3.22e-07 *** | ||
| + | #c 0.0222537 0.0054435 4.088 0.000766 *** | ||
| + | #d 0.2392522 0.3978514 0.601 0.555535 | ||
| + | |||
| + | #Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1 | ||
| + | |||
| + | #Residual standard error: 0.004108 on 17 degrees of freedom | ||
| + | |||
| + | #Number of iterations to convergence: 9 | ||
| + | #Achieved convergence tolerance: 1.49e-08 | ||
| + | plot(x, y, lwd=2, xlab='AuCl4_uM', ylab='Relative_bGal') | ||
| + | xfit <- seq(0, 10, 0.01) | ||
| + | yfit <- (0.0001397+0.041717/(1+exp(4*0.0222537*(0.2392522-xfit)/ 0.0434417+2))) | ||
| + | lines(spline(xfit, yfit), lwd=2) | ||
| + | |||
| + | #The effect of gold concentration | ||
| + | iGEM<-function(t, state, parms) { | ||
| + | with(as.list(c(state, parms)), { | ||
| + | |||
| + | if(Uin<0) {Uin<-0} | ||
| + | else {Uin<-Uin} | ||
| + | if(Uout<0) {Uout<-0} | ||
| + | else {Uout<-Uout} | ||
| + | |||
| + | dR <- (a + b/(1 + exp(4 * c * (d - Uin)/b + 2)))-R | ||
| + | dUin <- (e*(Uout-Uin)) | ||
| + | dUout <- (e*(Uin-Uout)) | ||
| + | |||
| + | list(c(dR, dUin, dUout)) | ||
| + | }) | ||
| + | } | ||
| + | |||
| + | library(deSolve) | ||
| + | |||
| + | parms<-c( | ||
| + | a=0.0001397, | ||
| + | b=0.0434417, | ||
| + | c=0.0222537, | ||
| + | d=0.2392522, | ||
| + | e=0.01) | ||
| + | |||
| + | times<-seq(0, 120, 0.05) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=0) | ||
| + | out0 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=0.5) | ||
| + | out1 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=1) | ||
| + | out2 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=2) | ||
| + | out3 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=4) | ||
| + | out4 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=6) | ||
| + | out5 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=8) | ||
| + | out6 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=10) | ||
| + | out7 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | |||
| + | out<-rbind(out0, out1, out2, out3, out4, out5, out6, out7) | ||
| + | plot(out, pch='.') | ||
| + | |||
| + | |||
| + | #The effect of membrane permeability | ||
| + | iGEM<-function(t, state, parms) { | ||
| + | with(as.list(c(state, parms)), { | ||
| + | |||
| + | if(Uin<0) {Uin<-0} | ||
| + | else {Uin<-Uin} | ||
| + | if(Uout<0) {Uout<-0} | ||
| + | else {Uout<-Uout} | ||
| + | |||
| + | dR <- (a + b/(1 + exp(4 * c * (d - Uin)/b + 2)))-R | ||
| + | dUin <- (e*(Uout-Uin)) | ||
| + | dUout <- (e*(Uin-Uout)) | ||
| + | |||
| + | list(c(dR, dUin, dUout)) | ||
| + | }) | ||
| + | } | ||
| + | time<-seq(0, 400, 1) | ||
| + | |||
| + | library(deSolve) | ||
| + | parms<-c( | ||
| + | a=0.0001397, | ||
| + | b=0.0434417, | ||
| + | c=0.0222537, | ||
| + | d=0.2392522, | ||
| + | e=0.01) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=0) | ||
| + | out0 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | times<-seq(0, 400, 0.1) | ||
| + | |||
| + | parms<-c( | ||
| + | a=0.0001397, | ||
| + | b=0.0434417, | ||
| + | c=0.0222537, | ||
| + | d=0.2392522, | ||
| + | e=0.01) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=5) | ||
| + | out1 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | parms<-c( | ||
| + | a=0.0001397, | ||
| + | b=0.0434417, | ||
| + | c=0.0222537, | ||
| + | d=0.2392522, | ||
| + | e=0.05) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=5) | ||
| + | out2 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | parms<-c( | ||
| + | a=0.0001397, | ||
| + | b=0.0434417, | ||
| + | c=0.0222537, | ||
| + | d=0.2392522, | ||
| + | e=0.005) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=5) | ||
| + | out3 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | |||
| + | out<-rbind(out0, out1, out2, out3) | ||
| + | plot(out, pch='.') | ||
| + | |||
| + | |||
| + | #The effect of gold mineralization peptides | ||
| + | #mineralisastion to peptide degradation ratio | ||
| + | iGEM<-function(t, state, parms) { | ||
| + | with(as.list(c(state, parms)), { | ||
| + | |||
| + | if(Uin<0) {Uin<-0} | ||
| + | else {Uin<-Uin} | ||
| + | if(Uout<0) {Uout<-0} | ||
| + | else {Uout<-Uout} | ||
| + | if(Uout==0) {P<-0} | ||
| + | else {P<-P} | ||
| + | |||
| + | dR <- (a + b/(1 + exp(4 * c * (d - Uin)/b + 2)))-R | ||
| + | dUin <- (e*(Uout-Uin)) | ||
| + | dUout <- (e*(Uin-Uout)-f*P) | ||
| + | dP<-(g*R/(h+R)-i*P) | ||
| + | |||
| + | list(c(dR, dUin, dUout, dP)) | ||
| + | }) | ||
| + | } | ||
| + | |||
| + | library(deSolve) | ||
| + | |||
| + | times<-seq(0, 150, 0.1) | ||
| + | |||
| + | parms<-c( | ||
| + | a=0.0001397, | ||
| + | b=0.0434417, | ||
| + | c=0.0222537, | ||
| + | d=0.2392522, | ||
| + | e=0.1, | ||
| + | f=0.1, | ||
| + | g=0.1, | ||
| + | h=0.1, | ||
| + | i=0.1) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=0, | ||
| + | P=0) | ||
| + | out0 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | state<-c( | ||
| + | R=0.003464946, | ||
| + | Uin=0, | ||
| + | Uout=5, | ||
| + | P=0) | ||
| + | |||
| + | out1 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | parms<-c( | ||
| + | a=0.0001397, | ||
| + | b=0.0434417, | ||
| + | c=0.0222537, | ||
| + | d=0.2392522, | ||
| + | e=0.1, | ||
| + | f=0.12, | ||
| + | g=1, | ||
| + | h=1, | ||
| + | i=0.1) | ||
| + | out2 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | parms<-c( | ||
| + | a=0.0001397, | ||
| + | b=0.0434417, | ||
| + | c=0.0222537, | ||
| + | d=0.2392522, | ||
| + | e=0.1, | ||
| + | f=0.2, | ||
| + | g=1, | ||
| + | h=1, | ||
| + | i=0.1) | ||
| + | out3 <- ode(y = state, times = times, func = iGEM, parms = parms) | ||
| + | |||
| + | out<-rbind(out0, out1, out2, out3) | ||
| + | plot(out, pch='.') | ||
| + | </code></pre></div> | ||
| + | <div id="labbookcontent" class="hiddenContent"><p><h2>August 22nd</h2></p> | ||
| + | <p>Assembly of the LuxR generator and the AiiA (BBa_K516022). We cleaved each part with XbaI and PstI (LuxR generator) or EcoRI and SpeI (AiiA). They were run on 1.2% agarose gels and extracted using QIAquick gel extraction kit. Through three way ligation, we managed to clone both inserts into the BBa_J61002 backbone that was precut with with EcoRI and PstI. We confirmed the insertion by amplifying the ligation products with GoTaq polymerase. The shift to 2Kb in lane D indicates the successful ligation reaction. We did transformations with <i>E. coli</i> strain DH5 alpha, and grew them on LB agar supplemented with 100ug/ml ampicilin.</p> <p><img src="https://static.igem.org/mediawiki/2013/4/46/Gelone.jpg" width="40%"><br clear=all></p> | ||
| + | <h2>August 23rd</h2> | ||
| + | <p>We performed colony PCR to screen for the transformants. After 12 hours of incubation, colonies were small and appeared white, so we left them to grow for three more hours. Most colonies appeared to have the right plasmid as they remained unchanged. 14 white colonies were picked at random and used for the PCR. We observed the same band shift which is positive.</p> <p><img src="https://static.igem.org/mediawiki/2013/9/9e/Geltwo.jpg" width="40%"><br clear=all></p> | ||
| + | <h2>August 24th</h2> | ||
| + | <p>We tried to assemble the gold sensing device (BBa_K1127008) with the LuxR/AiiA generators. Unfortunately, it was not successful. We think that the digestion could have been incomplete as bands were observed in the controls.</p> <p><img src="https://static.igem.org/mediawiki/2013/d/d0/Gelthree.jpg" width="40%"><br clear=all></p> | ||
| + | <h2>August 27th</h2> | ||
| + | <p>Today, we tried to the assembly again. Instead of the three way ligation, we used the Rapid Assembly of Biobricks (RAB). The parts were amplified with Phusion ® polymerase and digested by single restriction enzyme (SpeI for the BBa_K1127008, and XbaI for the LuxR/AiiA generators). The digests were ligated and subjected to PCR amplification. The band is shifted in lane A that indicates successful ligation. </p><p><img src="https://static.igem.org/mediawiki/2013/a/aa/Gelfour.jpg" width="40%"><br clear=all></p> | ||
| + | <h2>August 28th</h2> | ||
| + | <p>We returned to our favourite cloning techniques - the three way assembly. This time we succeeded. For confirmation, we amplified the ligation products. The shift to about 1.5Kb is shown in the gel photo.</p> <p><img src="https://static.igem.org/mediawiki/2013/a/a8/Gelfive.jpg" width="40%"><br clear=all></p> | ||
| + | <h2>September 9th</h2> | ||
| + | <p>For the purpose of characterization, we cloned a reporter gene (GFP or RFP generator) into our construct. Using the three way ligation, we managed to do it successfully. The gel photo shows how we cut off the gels for extraction. We also show the streaked colonies of the transformants. They are green which indicates the presence of GFP (DLig 13E).</p> <p><img src="https://static.igem.org/mediawiki/2013/9/9d/Gelsix.jpg" width="40%"><br clear=all></p><p><img src="https://static.igem.org/mediawiki/2013/0/0f/Gelseven.jpg" width="40%"><br clear=all></p> | ||
| + | |||
| + | </div> | ||
| + | <div id="niklabbookcontent" class="hiddenContent"><p><h2>Nikola's lab book</h2></p><p>His lab book can be found online <a href="https://static.igem.org/mediawiki/2013/8/8f/Nikola%27s_lab_book.pdf">here.</a> | ||
| + | |||
| + | </div> | ||
| + | |||
</div> | </div> | ||
Latest revision as of 14:47, 21 October 2013

July 10th
Purification of genomic DNA from Shewanella oneidensis and Salmonella sp.
July 25th
After a long break we finally started working on the actual assembly of parts - the mtr complex. Today Jonas ran a trial PCR on mtrB and mtrC part of the complex both of which are around the size of 2kb. GoTaq polymerase (Promega) was used, this is the polymerase we generally use for trials and colony PCRs as fidelity is not the main priority.
 This is the gel from the PCR. Temperatures ranged 46-71˚C. It appears that the primers are sufficiently specific at even low temperatures. 1% agarose, SYBR-Safe(Invitrogen), QStep4 ladder.
This is the gel from the PCR. Temperatures ranged 46-71˚C. It appears that the primers are sufficiently specific at even low temperatures. 1% agarose, SYBR-Safe(Invitrogen), QStep4 ladder.
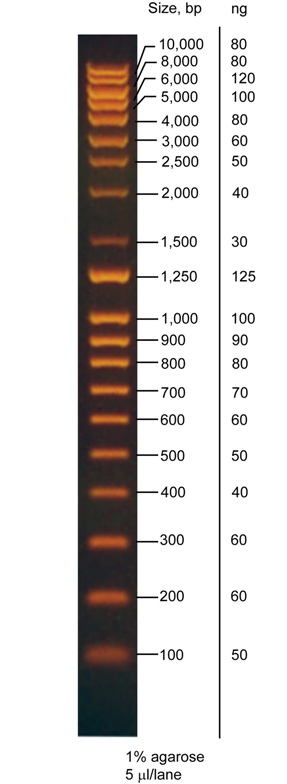{kind=link}
July 26th
Trial PCR again, this time for three small (~500bp) synthetic DNA fragments - gBlocks (IDT), these fragments are highlighted below on the diagram as well as the overall view of our mtr gene.

 The gel. Three different temperatures (51-61˚C)- all work. 1% agarose dyed with SYBR-Safe, however, due to poor practice the ladder is indistinguishable.
The gel. Three different temperatures (51-61˚C)- all work. 1% agarose dyed with SYBR-Safe, however, due to poor practice the ladder is indistinguishable.
At this point Jonas arrived at the realisation that as long as the primers are specific enough, there is no point in doing PCR trials (silly Jonas). Therefore most of the PCRs done this day forth use a universal 58˚C temperature.
July 27th
This is the first day that we actually got some positive outcome from our cloning. Or so we thought.
All the fragments of our mtr complex were once again amplified via PCR but this time using Phusion polymerase for high fidelity (Thermo Fisher). Gel confirmed that everything went fine and specifically. Using QIAquick PCR purification kit (Qiagen) we clean up the PCR product, then measure the concentrations using NanoDrop1000, do the required calculations and then proceed to the main reaction:
In 60 minutes at 50 ˚C we joined all of the 5 fragments using the Gibson Assembly Master Mix provided by NEB. Confirmed by a PCR.
 The PCR was not fully specific but it is unclear whether these fragments formed during Gibson reaction (main fragment joining sites are identical RBS) or because the temperature used was too low (51˚C & 58˚C). 3rd and 4th lanes are negative controls. 1% agarose dyed with SYBR-Safe, QStep4 ladder.
The PCR was not fully specific but it is unclear whether these fragments formed during Gibson reaction (main fragment joining sites are identical RBS) or because the temperature used was too low (51˚C & 58˚C). 3rd and 4th lanes are negative controls. 1% agarose dyed with SYBR-Safe, QStep4 ladder.
Before August 11th
During this time we tried putting all of our genes inside the pSB1C3 iGEM plasmid but with little luck. Everything seemed to be working fine up until the point when we realised that colony PCRs are not so trustworthy. It appeared clear that most of the colony PCRs came out as false positives after we decided to run the PCR on already purified plasmids. However, some samples were already on their way for sequencing (thinking that we actually got something within our pSB1C3 backbones we sent them). The sequencing results gave us an answer: we planned it all wrong! Graphical summary:

So all of our parts that were to be put inside the iGEM plasmid only had XbaI and SpeI cutting sites, but we were unaware that they actually had complementary sticky ends and it was nearly impossible to ligate something in between.
We fixed it with new primers which add the remaining part of the prefix and suffix. However, we lost 2 weeks worth of productive labwork.
August 12th
So after receiving the new primers we continue working. This time we're using Gibson's assembly to directly insert our mtr part into pSB1C3, this allows us to skip any problems that might occur during digestion or ligation.Note that during design of these primers we decided to produce two different mtr parts, one of which would lack the promoter and the first RBS(crR12) which we thought might not be as convenient for other teams.
So today we tried amplifying mtrAB(from the previous Gibson), mtrC(from previous PCR, should have used the genome) and pSB1C3 backbone(the linearised plasmid from iGEM HQ). Unfortunately, the backbone PCR failed.
 We thought that perhaps dimers of the primers formed as the sequences of prefix and suffix are not that different. 1% agarose dyed with SYBR-Safe, 1kb ladder(Invitrogen)(on the right).
We thought that perhaps dimers of the primers formed as the sequences of prefix and suffix are not that different. 1% agarose dyed with SYBR-Safe, 1kb ladder(Invitrogen)(on the right).
{kind=link}
However, after playing a bit with DMSO and annealing temperatures we were still unable to amplify it until we changed the template. We took a random iGEM part we found in the fridge and tried the PCR on it and it worked. Conclusion: iGEM HQ should provide more information about their plasmid linearisation and how were the ends modified.
August 13th
2nd and 3rd Gibson Assembly. So we purified the assembly parts from previous PCR and did the calculations to maximise the amount of product. This reaction was also relatively simple as we reduced the number of fragments to 3 and it results in already circular plasmid.
 So while we do the transformations, spending some usual late time in the labs, we also did a PCR using VF2 and VR primers. The gel confirms that reactions were successful. We can see the bands just above the 5kb mark,and as there is only about 130bp difference between these distinct mtr constructs it is impossible to see that on the gel. 1% agarose dyed with SYBR-Safe, 1kb ladder(Invitrogen)(on the right).
So while we do the transformations, spending some usual late time in the labs, we also did a PCR using VF2 and VR primers. The gel confirms that reactions were successful. We can see the bands just above the 5kb mark,and as there is only about 130bp difference between these distinct mtr constructs it is impossible to see that on the gel. 1% agarose dyed with SYBR-Safe, 1kb ladder(Invitrogen)(on the right).
August 14th
So after having some sleep we head back to the lab and do the colony PCR on our transformations. But because Jonas is a very lazy person he used Phusion polymerase (which is as much as 4 times faster than GoTaq) and failed - there is nothing visible on the gel apart from ladder. It is not the first time this happens with Phusion colony PCR, so we try to avoid doing it, it appears though that Phusion requires relatively pure DNA template.
Nonetheless, we inoculate 2 cultures each of both mtr ORFs only and mtr+promoter constructs and grow them overnight. But it is not all for the day.
Also, today we got the new primers for the rest of the cloning, so we amplified the sequences of gold peptides and gold sensing device adding the rest of prefix and suffix. It all worked but the gel is really really hideous thus it will not appear up in here. We leave the rest for tomorrow.
August 15th
We extract the plasmids from overnight cultures using NucleoSpin Plasmid(Macherey-Nagel) and digest them with ThermoFisher's FastDigest enzymes:
 Every triplet is: 1st the control, 2nd PstI digest and 3rd the EcoRI & SpeI digest. It is obvious that there is nothing near the size of 5kb visible. However, the 1kb fragment that is visible in double digest shows us that it is actually the template we used for pSB1C3 backbone amplification. 1% agarose dyed with SYBR-Safe, 1kb ladder(Invitrogen)(on the right).
Every triplet is: 1st the control, 2nd PstI digest and 3rd the EcoRI & SpeI digest. It is obvious that there is nothing near the size of 5kb visible. However, the 1kb fragment that is visible in double digest shows us that it is actually the template we used for pSB1C3 backbone amplification. 1% agarose dyed with SYBR-Safe, 1kb ladder(Invitrogen)(on the right).
So the solution is simple: we digest the Gibson reactions we had with DpnI(Promega) thus removing the background and then repeat the transformations.
At the same time we continued working with other sequences. Amplified gBlocks (peptides,the gold device) were purified with QIAquick kit, digested with EcoRI & PstI and then purified again using the same kit - this step frees our samples of small 2-6bp long fragments making the ligation easier. We also digest the linearised plasmid from iGEM HQ with the same enzymes and purify it as well. Even though this leads us to a very low concentration of pSB1C3, it is still enough for transformations as long as there are no problems with ligations. So we then make the reactions with T4 Ligase(Thermo Fisher) and proceed with the transformations.
August 16th
The day of GoTaq colony PCRs. So basically Gintare marched to the lab to do PCR of 44 colonies (and we are all aware of how annoying it is). Results were pretty straightforward with the peptides and gold sensing device parts. On the left gel below first four are BBa_K1127000, then BBa_K1127001 and BBa_K1127003. On the second gel: BBa_K1127002, followed by two constituents (which we join together later) of BBa_K1127008 and then BBa_K1127010 which was not submitted. Two colonies of each were cultivated overnight.


However, the results for mtr complexes were weird. So we picked a few cultures and grew them overnight for further analysis.


All gels were 1% agarose dyed with SYBR-Safe, 1kb ladder(Invitrogen)(on the right). We also noticed that something was not right with our gel viewing box as the images turned out to be very dark even at max exposure.
August 17th
Another busy day, so firstly about mtr complex. We purified the plasmids from yesterdays colonies using our usual NucleoSpin Plasmid kit and then digest them with FastDigest enzymes:

Triplet consists of: 1. Control 2. PstI digest 3. EcoRI & SpeI digest. First three triplets are of the mtr ORFs only construct and the last one (a lot of it!) is the one with promoter. Even though the gel is faint we can see the most important thing: double digests separate the pSB1C3(2kb) backbone and our part (5.3kb). The PstI digest also confirms the prohibited PstI cutting site within mtrC sequence (present in the genome). 1% agarose dyed with SYBR-Safe, 1kb ladder(Invitrogen)(on the right).
Using the primers we designed earlier we do the Site-Directed Mutagenesis on each of the mtr constructs, which should remove the PstI cutting site from where it should not be present but still retaining the aminoacid sequence. We also finish it up with DpnI digestion to remove any background and then transform the cells.
In the meantime, we did similar to mtr digestions on the peptide parts we cloned earlier. Again on NucleoSpin purified plasmids with FastDigest enzymes.


Each plasmid was ran on the gel in triplets: 1. uncut control 2. XbaI & SpeI digest 3. EcoRI & PstI digest. Both gels show the same peptides mentioned earlier from different colonies picked yesterday. The gel shows us that the peptides are actually there within the pSB1C3 with working prefixes and suffixes (there was only a slight nuisance with the 3rd peptide). 1% agarose gels dyed with either ethidium bromide(left) or SYBR-Safe(right), QStep4 ladder.
August 18th
Today we do colony PCR (yet again!) on our SDM'd plasmid. A clever way we thought of to check if PstI cutting site is still there was to amplify the mtrC part of the colony and digest it immediately with PstI:
 Although it was not so cleverly thought to the end: Jonas forgot the control for mtrC before the SDM. Basically, the first pair is the control on the SDM reagent from yesterday, the rest of them are our mtr constructs. First member of the pair is undigested product and the other one is PstI digested product. We can see the the bands are not at the equal sizes, however, that's an optical illusion caused by different volumes added to the lanes (yes, we checked under the manual UV lamp). 1% agarose dyed with SYBR-Safe(right), QStep4 ladder(although irrelevant).
Although it was not so cleverly thought to the end: Jonas forgot the control for mtrC before the SDM. Basically, the first pair is the control on the SDM reagent from yesterday, the rest of them are our mtr constructs. First member of the pair is undigested product and the other one is PstI digested product. We can see the the bands are not at the equal sizes, however, that's an optical illusion caused by different volumes added to the lanes (yes, we checked under the manual UV lamp). 1% agarose dyed with SYBR-Safe(right), QStep4 ladder(although irrelevant).
We also tried joining the two mentioned parts to produce BBa_K1127008 using Amplified Insert Assembly.
August 19th
And it did not work for us.
 This is the colony PCR with VF2&VR primers and all the bands are clearly lower than the expected 900bp size, it actually fits the size of our recipient plasmid. 1% agarose dyed with SYBR-Safe, Hyper ladder 1kb(Bioline).
This is the colony PCR with VF2&VR primers and all the bands are clearly lower than the expected 900bp size, it actually fits the size of our recipient plasmid. 1% agarose dyed with SYBR-Safe, Hyper ladder 1kb(Bioline).
{kind=link}
We spent the remaining time designing primers for the sequencing of whole mtr complex.
August 22nd
After trying a few other techniques and failures of joining our two fragments together (note that they are now in biobrick format) we decided to test something new. Basically we joined the parts together before putting them inside the plasmid:

August 23rd
We select two random colonies from yesterday transformations and grow them overnight.
Also, we receive sequencing primers for mtr and send the samples to Source BioScience.
August 24th
Purified plasmids from cultures are cut to confirm that we have the gold sensing particle ready.
 Left and right of the marker are two plasmids: first the uncut control, XbaI digest, SpeI digest, EcoRI&PstI double digest followed by SanDI digest. Double digest reveals the ~750bp long fragment which proves our method successful. 1% agarose dyed with SYBR-Safe, Hyper ladder 1kb(Bioline).
Left and right of the marker are two plasmids: first the uncut control, XbaI digest, SpeI digest, EcoRI&PstI double digest followed by SanDI digest. Double digest reveals the ~750bp long fragment which proves our method successful. 1% agarose dyed with SYBR-Safe, Hyper ladder 1kb(Bioline).
August 27th
We receive our first sequencing results of mtr complexes. Unfortunately, we found a single base pair substitution at the very start of mtrA (changing the aminoacid lysine to glutamic acid) and a nonsense mutation in mtrC. However, the mtr construct with promoter contained an additional nonsense mutation within mtrA, this is the point where we decided to give up on this one and try and fix the other, more sensible biobrick.
So we ignore the single base pair substitution (as it is not part of the protein motive compared with other mtrA analogs) and order new set of primers for the Site-Directed Mutagenesis of mtrC.
August 30th
We receive the primers for mtr SDM from IDT and do it immediately the same way we did it last time.
Samples for sequencing of the peptides and gold sensing constructs were sent today.
September 4th
We do the β-galactosidase assay on the gold sensing device for which the results can be found elsewhere.
The mtr part was sent for sequecing again to see if the SDM was successful.
September 10th
Finally, DNA of the parts for submission is prepared and is finally sent to iGEM HQ on September 16th.
February 21st
Today the iGEM team divided to discuss about the two main ideas, chosen from over 100, after meeting with potential supervisors.
The first idea is making a mouthwash which contains harmless, live bacteria which normally reside in the mouth. These bacteria will be genetically engineered to secrete pleasant odours (e.g. menthol, rose, banana - parts for which have already been submitted to the iGEM registry). The scope is to have the bacteria colonise the mouth; to prevent the smell being secreted at all times, their production could be controlled by controlling gene expression, depending on external signals (such as concentration of oxygen, glucose or stress).
Becca, Evaldas, Andy, Jonas, Andra, Hannah and Caroline looked into the idea in more depth. One of the major concerns is that safety issues would prevent the mouthwash from being regarded as a product which can be used, since it involves introducing live, genetically modified bacteria into humans. Also, the idea is not conceptually very challenging.
The second idea is to create an improved microbial fuel cell, a type of battery that uses live bacteria for current production. The bacteria releases H ions and electrons into the environment, when encapsulated in an anode chamber. These secreted particles contribute to energy formation, when electrons pass from the anode to the cathode of the battery. We would like to genetically engineer E.coli to secrete more electrons, by introducing the secretion system present in a different organism, Shewanella oneidensis. This has been attempted in iGEM before, but never achieved. In addition, we would try to deposit gold nanoparticles on the anode of the battery, to increase its conductivity.
Ivan, Kobchai, Nick, Ric, Gintare, Rudolfs and Alex looked into this idea. The main challenges are that, although nanoparticles have been created before, we are unsure which genes we would use for this process. More reading through literature is needed to identify them, before carrying on.
February 27th
Each subteam presented their latest findings for the two ideas. Kobchai went to discuss with people from the engineering department, who gave useful insight into how nanoparticles are made in vivo and which organisms and gene could be used. We found out that Salmonella enterica has a good gold-detection system, which we could use to control gene expression in the presence of gold ions. We have also identified a few papers which describe how bacteria secrete nanoparticles. We decided this would be a good starting point to carry on with the gold idea.
We have given ourselves two weeks to divide work into subgroups and do reading on several issues; 1st year students will be grouped with older students, to ensure they receive support in interpreting data and understanding the articles. To do list:
● what genes would be involved in electron secretion?
● what method of nanoparticle formation would we use?
● how would we control gene expression?
● are there any parts in the iGEM Registry we could use?
March 13th
Ivan gave a comprehensive presentation on our findings so far, in front of potential supervisors. We have established that the main objective of our project is to improve existing microbial fuel cell technology and make fuel cells more cost effective. The three aims which lead to this are:
1) Expressing the MTR protein complex from Shewanella on the surface of E.coli for electron transport;
2) Creating a MFC of approx 3cm diameter and engineer E.coli to deposit gold nanoparticles on the anode;
3) Use a quorum sensing system to control gene expression of genes involved in gold deposition.
Ideas which emerged during the meeting concerned the following:
● would the electron transporter introduced affect bacterial metabolism?
● would the bacteria form biofilms on the anode? Could this be avoided by expressing pilli?
● what is the gold concetration needed and where would we take it from? Possible solution: pharmaceutical waste rich in gold ions, which would also allow the safe removal of toxic ions from the environment
● other metals have been discussed, as alternatives to gold, such as copper and silver. However, there are already genes which are involved in copper metabolism in bacteria, so their effects might interfere with our system.
● two bacterial strains could be used, living together inside the MFC, one for the electron secretion and one for the quorum sensing
May 1st
After the spring holiday, during which the team has worked from home on iGEM, we had a n important meeting with our supervisors.
We are considering using a peptide which can bind elemental gold, such as MIDAS, to create nanoparticles. We have discussed how to ensure the nanoparticles are deposited on the anode. There were several variants proposed:
● fuse the peptide to secreted protein and let the complex settle on the anode (by gravity or magnetism)
● fuse the peptide to surface proteins (such as OpmA) and then as above
● turn the whole chamber into a graphyte anode, to increase the chance of bacteria being trapped in the porous material and release nanoparticles there
● lyse bacteria once they have captured gold ions internally, to release the peptide, and then repopolate the MFC with the second bacterial strain, which secretes the MTR complex
● aqua regia was proposed as a source of gold ions for the initial stages of the project, as an alternative to pharmaceutical waste; the composition of the latter may vary between experiemtns and will prevent us from making controlled, accurate measurements between replicate experiments
May 15th
During today's meeting we discussed more the details of the peptides we want to create. We have looked into several secretion tags which we would like to fuse to the peptide. We have narrowed it down to two, for now. HlyA can transport small peptides outside bacteria (though our initial in silico simulations show the peptidic construct would not be very stable). We might need a special E.coli strain if we work with it. pelB is good for small peptides, easy to use and we have an available vector for it. Another option is to have a poly-peptide, made with the A3, nanoparticle-producing peptide, rather than MIDAS. This poly-A3 would be secreted and cleaved outside of the bacteria; this method would give more stability to the system.
Need to look more into:
● Physiology of bacteria in the MFC
● What we will compare (e.g. two MFC, one improved and one normal)
● We need to design a good, simple end-assay, which will allow us to test the effect of our peptide on electricity production even if the rest of the system does not work/is not ready yet (e.g., he suggests putting the peptide in Shewanella, if we're not ready closer to the finish, and show it produces NP in Shewanella).
June 5th
After exams, we divided into four groups and started doing extensive reading on genes, to start preparing the gene sequences we need to order. Ric, Hannah, Becca and Andra are looking into the gold sensing system existing in Salmonella, GolTS. Alex, Evaldas and Jonas are looking into peptide design and secretion. Ivan, Nick and Andy are looking into modelling and identifying genes in the iGEM Registry. Kobchai, Caroline and Rudolfs are looking into the quorum sensing system and system regulation as a whole.
Notable things so far: some of the sequences for the pGolTS system submitted by other teams to the Registry seem to be wrong; we need to look more into it during the following week.
We might use some of the parts from team Edinburgh, which were not submitted last year, but which they can send to us, for the mtr complex.
June 12th
Evaldas, Kobchai and Andra gave a presentation on what has been done so far. Andy talked about the modelling he worked on, using the kinetic values obtained from scientific papers. . We need to look more into the details of the MFC we are planning on making, in particular what buffers we are planning on keeping the bacteria in, whether we will grow the bacteria in an aerobic or anaerobic environment and what bacterial strains to choose.
June 19th
The team spent the week designing some of the constructs, but also looking into the details of the MFC construction. We came together to make some decisions regarding the latter. We have established that, to allow the bacteria to create biofilms, it would be better to grow them in an anaerobic environment in the final MFC. However, for the initial experiments, we will use an aerobic environment. The two bacterial strains we will use are K12 and W3110, as they have been used with MFC before, are well characterised and easily available.
We also met Prof. Smith, to discuss about methods of isolating the peptide once it is produced, for part characterisation. We chose to put a flag tag on the peptide, to be identified with antibodies by western blot.
June 26th
During our supervisory meeting we talked about which vectors are best to be used for our constructs, and which constructs should be put on what vectors. We have discussed the advantages of pET vs PUC vectors and other possibilities proposed. We decided to go with the 3 vectors of the pET duet, which have different antibiotic resistance and have compatible origins of replication. We would use one for the gold recognition, one for mtr and one for the quorum sensing.
We also showed the supervisors our defined sequences, hoping we will soon order the sequences. We presented the techniques we are planning to use for every part, to characterise it (beta galactosidase assays). Also, the methods of assembly were presented to the supervisors (amplified insert assembly, which they were unfamiliar with, and the formerly proposed Gibson assembly). We received positive feedback on our choices.
An issue was raised regarding the way in which we would isolate peptides from cells, to assess their presence (since they are too small to be run on a gel). We chose to perfor, pull down assays. Defining sequences. To be expanded.
July 2nd
We had a small discussion with Dr. Thomas, about ordering the sequences. We went over all the designed sequences, verified restriction sites and are now preparing to submit the online order for the sequences. We have received the ordered consumables and are now ready to start working in the lab. Our meetings from now on will be administrative meetings and troubleshooting meetings.
July 16th
Internal meeting to redistribute the work within the team
July 24th
Final meeting with Dr. Chong before he leaves for two weeks. Catch up with supervisors on how lab work is going.
Rapid Assembly of Biobricks (RAB)
We found this easy way of joining two or more biobricks together:
1. Amplify the parts using VF2&VR primers (although specific primers make it easier).
2. Digest one product with SpeI and the other one with XbaI.
3. We would usually gel extract the fragments we need, which makes the next step easier.
4. Ligate the two fragments together.
5. Now amplify with PCR again the whole fragment that should have formed.
6. You can continue joining more parts together by repeating the above steps.
7. Finally, insert your new part into the iGEM plasmid using the standard procedure.
8. Transform.
Now many would argue that this continuous PCR would result in lots of mutations. However, polymerases with already high fidelity are being continuously improved to the point where their mutation rate is hard to detect statistically. Also, we sequence the parts anyway, so there is high potential in this assembly. We successfully joined our gold sensing device using this protocol. If you feel confused about the procedure, perhaps this picture will help to clarify the mechanism:
Beta galactosidase assay for gold induction
Growth:
1. Transfer one colony to LB with antibiotics and grow overnight at +37 °C with shaking.
2. Next morning transfer bacteria to fresh LB with antibiotics up to OD600=0.1 and continue growing until exponential stage (OD600=0.6-0.8).
3. Induce with required concentration of AuCl4 and continue growing at 30 °C or 37 °C depending on the experiment.
4. Samples for measurements can be taken at time points or after overnight growth.
Measurements:
1. Take 200 µl of cell suspension and measure OD630 (due to machine that we used instead of OD600) in a 96 well plate.
2. Take 100 µl of cell suspension for beta gal assay and mix with 100ul Z-buffer.
3. Add 10 µl 0.1% SDS and 20 µl chloroform and incubate at 30 °C for 5 min to disrupt the cells.
4. Add 50 µl 5mM PNPG and incubate at 30 °C for 30 min (incubation times can be extended up to 1h if the colour is not appearing, however the same incubation time should be applied for all of the related samples).
5. To stop the reaction and increase the intensity of yellow colour add 100 µl of 1 M Na2CO3 and centrifuge and 13k rpm for 5 min.
6. Measure the absorbance at 405 nm.
7. The relative response (∅) was calculated according to the formula:
![]()
Reagents:
Z-buffer stock solution:
4.27 g Na2HPO4
2.75 g NaH2PO x 4H2O
0.375 g KCl
0.125 g MgSO4 x 7H2O
Adjust to pH 7.0.
Bring to 500 ml with dH2O. Do not autoclave. Store at 4°C.
For complete Z-buffer - Prior to daily use mix:
50 ml Z-buffer
0.14 ml ß-mercaptoethanol
1 M Na2CO3 (store at 4°C)
5.3 g Na2CO3
50 ml dH2O
Bacterial growth media recipes:
Luria-Bertani media
10g Tryptone
5g Yeast Extract
10g NaCl
(15g Agar for solid media)
dH2O to 1000ml. Autoclave.
M9 minimal media
6g NaHPO4
3g KH2PO4
0.5g NaCl
Mili-Q water to 975ml. Autoclave.
Under sterile conditions:
1ml 1000x vitamins
1ml 1000x trace metals
2ml 1M MgSO4 (autoclaved)
0.2ml 1M CaCl2 (autoclaved)
1ml 1000x antibiotic
20ml 20% glucose (filtered via 0.2 micron filter)
Preparation of chemically competent cells
Solutions:
TFB1:
30mM Potassium Acetate
10mM CaCl2
50mM MnCl2
100mM RbCl
15% glycerol
Adjust pH to 5.8. Filter sterilise (0.2µm). Store at room temperature.
TFB2:
10mM MOPS
75mM CaCl2
10mM RbCl
15% glycerol
Adjust pH to 6.5. Filter sterilise (0.2µm). Store at room temperature.
Notes:
Warm clothing is recommended for day 3.
Prechill all equipment to be used in the cold room.
When working from day 3 (step 4) keep the cells cold at all times and perform re-suspensions in the cold room.
Make sure to read the protocol thoroughly before starting.
Method:
1. Streak a non selective LB plate with a dilution of the previous batch of Competent Cells, incubate at 37°C overnight.
2. The next day, inoculate a single colony from the plate in 2.5ml of LB broth. Incubate at 37°C overnight with shaking at approximately 225rpm.
3. The next day, use the entire overnight culture to inoculate 250ml of LB medium containing 20mM MgSO4 (a 1/100 dilution of 2mM stock). Take 1ml of the culture and keep in a cuvette, as blank for OD measurement. Grow the cells in an autoclaved 1L flask until the OD600 reaches 0.4-0.6 (check after 2 hours then keep testing until within the range). A 1L flask is necessary for proper aeration and growth.
4. Pellet the cells by centrifugation at 4500g for 5 minutes at 4°C. For a 250ml culture use two 250ml centrifuge bottles in a large rotor (eg. Beckman), or if not available use 50ml tubes in a bench top centrifuge at 3500rpm for 15 minutes at 4°C. Tubes need to be prechilled!
5. Re-suspend the cell pellets in 0.4 volume (based on the original culture volume) of ice cold TFB1, using vortex, in the cold room. For a 250ml subculture, use 100ml of TFB1 (50ml/bottle). Combine the re-suspended cells in one bottle. For the remaining steps, keep the cells on ice and chill all pipettes, tubes and flasks.
6. Incubate the re-suspended cells on ice for 5 minutes at 4°C.
7. Pellet the cells by centrifugation as per step 4. When transporting cells to centrifuge, keep them on ice.
8. Re-suspend the cells in 1/25 of the original culture volume of ice cold TFB2, using vortex, in the cold room. For a 250ml subculture use 10ml of TFB2.
9. Incubate the cells on ice for 60 minutes and then aliquot 100µl/ Eppendorf tube for storage at -80°C.
10. Quick freeze the tubes in a dry ice/isopropanol bath (or liquid nitrogen). Cells should be stable for between 3 months to 1 year.
Transformation:
1. Thaw the competent cells on ice. Use 50µl per transformation.
2. Gently mix the cells with up to 5µl of DNA without pipetting up and down.
3. Incubate on ice for 20 minutes.
4. Heat shock the cells at 42°C for 45s.
5. Place the cells on ice for 3-5 minutes.
6. Add 200-950µl of room temperature LB or SOC media.
7. Incubate the cells at 37°C whilst shaking them at 225rpm for 1hour (2 hours if plasmid contains chloramphenicol resistance).
8. Spread up to 200µl on the selection plates and incubate at 37°C overnight.
Colony PCR
1. Combine the master mix for PCR on ice (20µl reactions are sufficient).
2. Dip the pipet tip to the selected colony and now either:
a) Pipet up and down directly into PCR mix aliquot and store the tip inside eppie in the fridge, or
b) Pipet the bacteria into 10µl of H2O and pipet 1µl into the reaction, storing the rest in the fridge
3.Continue the PCR as normal and run the gel, you can then grow the selected cultures from the bacteria you saved in the fridge.
Site-Directed Mutagenesis
1. Design the primers with mutation that meet end to end (there is software for that) and have them synthesized 5' phosphorylated (SDS-PAGE purification recommended).
2. Perform a PCR as usual with high fidelity polymerase.
3. Recircularise the amplified plasmid with DNA ligase.
4. Digest with DpnI. This is optional if you use little amounts of template (as low as 10pg) and are going to send 3 or more samples for sequencing.
5. Transform.
Models for the gold sensing device (BBa_K1127008)
Solutions to this problem are relatively simple. The law of mass action which is widely used in biochemistry, physiology and pharmacology is one of the straightforward approaches. However, it requires a massive amount of data at different points in time. This is very tricky to maintain accuracy and to spend less resource. Fitting pre-existing models in our data came to us as a plausible solution. So we measured activities of the device when it reached steady states. Data were collected and fit into the nonlinear models for comparison.

We computed information Akaike Information Criterion (AIC) to determine quality of the models. Regardless of values of the goodness of fit, Logistic is the best model among the three. So model parameters of the Logistic are estimated and shown here.

The model is simulated and compared with the experimental data. As seen in the Figure 1, Logistic is well fit and could be a good reflection of reality.

The model is extended to include diffusion of the gold. 'e' is the diffusion constant. In the other word, it is the permeability of the bacterial envelope for the ion Au(III). We solved the differential equations and produced simulations to predict behaviour of the system in various conditions.

The model predicts multiple steady. As seen in the Figure 2, the system could reach higher steady states with increasing gold concentration.

In Figure 3, the system is fairly robust. It could approach the same steady state despite of differences in the diffusion constants.

For the sake of curiosity, we include expression of the synthetic peptide for gold bio-mineralization. Extension of the model is shown below.

Literature about this peptide is scarce, so we make following assumptions:
1.  is the change in expression of the peptide;
is the change in expression of the peptide;
2. Gold bio-mineralization is a linear process with the constant rate 'f';
3. Secretion of the peptide is in the form of standard Hill equation  with Hill coefficient=1;
with Hill coefficient=1;
4. The peptide is degraded through a linear process i[peptide]out.
The model is simulated and shown in Figure 4. It suggests that the system could undergo the decline phase. The rates of reduction could vary depending on the rates of bio-mineralization.

Our models can't produce precise numerical solutions of the real world problems. However, they have capabilities to describe structures and to generate broad outcomes to what might happen in certain conditions. This qualitative approach is actually advantageous as it provides guidance for experimental design and data analysis without overloading us with unnecessary information.
R Source code
#Model fitting
library(minpack.lm)
library(bbmle)
library(plyr)
library(ggplot2)
#Observed data between 0uM and 10uM
x<-c(0,0,0,0.5,0.5,0.5,1,1,1,2.5,2.5,2.5,5,5,5,7.5,7.5,7.5,10,10,10)
y<-c(0.002857143,0.00152381,0.00212963,0.011598174,0.009054726,0.011746032,0.021904762,0.00854902,0.016464646,0.038600823,
0.043166667,0.043508772,0.045142857,0.045367965,0.035365854,0.043474178,0.044122807,0.04971831,0.03822807,0.045063291,
0.043288889)
#0.043508772<=0.053508772, 0.04971831<=0.05971831, 0.03822807<=0.033?
df<-data.frame(x, y)
#HILL FUNCTION
Hill<-nlsLM(y~((a+b*x^d)/(c+x^d)), start=list(a=0.002, b=0.044, c=1, d=2), trace=TRUE)
#LOGISYIC FUNCTION
Logt<-nlsLM(y~(a+b/(1+exp(4*c*(d-x)/b+2))), start=list(a=0.001, b=0.044, c=0.025, d=0.1), trace=TRUE)
#GOMPERTZ FUNCTION
Gomp<-nlsLM(y~(a+b*exp(-exp(c*2.71828*(d-x)/b+1))), start=list(a=0.001, b=0.044, c=0.025, d=0.1), trace=TRUE)
AICtab(Hill, Logt, Gomp)
# dAIC df
#Logt 0.0 5
#Gomp 1.8 5
#Hill 3.6 5
summary(Logt)
#Formula: y ~ (a + b/(1 + exp(4 * c * (d - x)/b + 2)))
#Parameters:
# Estimate Std. Error t value Pr(>|t|)
#a 0.0001397 0.0048826 0.029 0.977507
#b 0.0434417 0.0053800 8.075 3.22e-07 ***
#c 0.0222537 0.0054435 4.088 0.000766 ***
#d 0.2392522 0.3978514 0.601 0.555535
#Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
#Residual standard error: 0.004108 on 17 degrees of freedom
#Number of iterations to convergence: 9
#Achieved convergence tolerance: 1.49e-08
plot(x, y, lwd=2, xlab='AuCl4_uM', ylab='Relative_bGal')
xfit <- seq(0, 10, 0.01)
yfit <- (0.0001397+0.041717/(1+exp(4*0.0222537*(0.2392522-xfit)/ 0.0434417+2)))
lines(spline(xfit, yfit), lwd=2)
#The effect of gold concentration
iGEM<-function(t, state, parms) {
with(as.list(c(state, parms)), {
if(Uin<0) {Uin<-0}
else {Uin<-Uin}
if(Uout<0) {Uout<-0}
else {Uout<-Uout}
dR <- (a + b/(1 + exp(4 * c * (d - Uin)/b + 2)))-R
dUin <- (e*(Uout-Uin))
dUout <- (e*(Uin-Uout))
list(c(dR, dUin, dUout))
})
}
library(deSolve)
parms<-c(
a=0.0001397,
b=0.0434417,
c=0.0222537,
d=0.2392522,
e=0.01)
times<-seq(0, 120, 0.05)
state<-c(
R=0.003464946,
Uin=0,
Uout=0)
out0 <- ode(y = state, times = times, func = iGEM, parms = parms)
state<-c(
R=0.003464946,
Uin=0,
Uout=0.5)
out1 <- ode(y = state, times = times, func = iGEM, parms = parms)
state<-c(
R=0.003464946,
Uin=0,
Uout=1)
out2 <- ode(y = state, times = times, func = iGEM, parms = parms)
state<-c(
R=0.003464946,
Uin=0,
Uout=2)
out3 <- ode(y = state, times = times, func = iGEM, parms = parms)
state<-c(
R=0.003464946,
Uin=0,
Uout=4)
out4 <- ode(y = state, times = times, func = iGEM, parms = parms)
state<-c(
R=0.003464946,
Uin=0,
Uout=6)
out5 <- ode(y = state, times = times, func = iGEM, parms = parms)
state<-c(
R=0.003464946,
Uin=0,
Uout=8)
out6 <- ode(y = state, times = times, func = iGEM, parms = parms)
state<-c(
R=0.003464946,
Uin=0,
Uout=10)
out7 <- ode(y = state, times = times, func = iGEM, parms = parms)
out<-rbind(out0, out1, out2, out3, out4, out5, out6, out7)
plot(out, pch='.')
#The effect of membrane permeability
iGEM<-function(t, state, parms) {
with(as.list(c(state, parms)), {
if(Uin<0) {Uin<-0}
else {Uin<-Uin}
if(Uout<0) {Uout<-0}
else {Uout<-Uout}
dR <- (a + b/(1 + exp(4 * c * (d - Uin)/b + 2)))-R
dUin <- (e*(Uout-Uin))
dUout <- (e*(Uin-Uout))
list(c(dR, dUin, dUout))
})
}
time<-seq(0, 400, 1)
library(deSolve)
parms<-c(
a=0.0001397,
b=0.0434417,
c=0.0222537,
d=0.2392522,
e=0.01)
state<-c(
R=0.003464946,
Uin=0,
Uout=0)
out0 <- ode(y = state, times = times, func = iGEM, parms = parms)
times<-seq(0, 400, 0.1)
parms<-c(
a=0.0001397,
b=0.0434417,
c=0.0222537,
d=0.2392522,
e=0.01)
state<-c(
R=0.003464946,
Uin=0,
Uout=5)
out1 <- ode(y = state, times = times, func = iGEM, parms = parms)
parms<-c(
a=0.0001397,
b=0.0434417,
c=0.0222537,
d=0.2392522,
e=0.05)
state<-c(
R=0.003464946,
Uin=0,
Uout=5)
out2 <- ode(y = state, times = times, func = iGEM, parms = parms)
parms<-c(
a=0.0001397,
b=0.0434417,
c=0.0222537,
d=0.2392522,
e=0.005)
state<-c(
R=0.003464946,
Uin=0,
Uout=5)
out3 <- ode(y = state, times = times, func = iGEM, parms = parms)
out<-rbind(out0, out1, out2, out3)
plot(out, pch='.')
#The effect of gold mineralization peptides
#mineralisastion to peptide degradation ratio
iGEM<-function(t, state, parms) {
with(as.list(c(state, parms)), {
if(Uin<0) {Uin<-0}
else {Uin<-Uin}
if(Uout<0) {Uout<-0}
else {Uout<-Uout}
if(Uout==0) {P<-0}
else {P<-P}
dR <- (a + b/(1 + exp(4 * c * (d - Uin)/b + 2)))-R
dUin <- (e*(Uout-Uin))
dUout <- (e*(Uin-Uout)-f*P)
dP<-(g*R/(h+R)-i*P)
list(c(dR, dUin, dUout, dP))
})
}
library(deSolve)
times<-seq(0, 150, 0.1)
parms<-c(
a=0.0001397,
b=0.0434417,
c=0.0222537,
d=0.2392522,
e=0.1,
f=0.1,
g=0.1,
h=0.1,
i=0.1)
state<-c(
R=0.003464946,
Uin=0,
Uout=0,
P=0)
out0 <- ode(y = state, times = times, func = iGEM, parms = parms)
state<-c(
R=0.003464946,
Uin=0,
Uout=5,
P=0)
out1 <- ode(y = state, times = times, func = iGEM, parms = parms)
parms<-c(
a=0.0001397,
b=0.0434417,
c=0.0222537,
d=0.2392522,
e=0.1,
f=0.12,
g=1,
h=1,
i=0.1)
out2 <- ode(y = state, times = times, func = iGEM, parms = parms)
parms<-c(
a=0.0001397,
b=0.0434417,
c=0.0222537,
d=0.2392522,
e=0.1,
f=0.2,
g=1,
h=1,
i=0.1)
out3 <- ode(y = state, times = times, func = iGEM, parms = parms)
out<-rbind(out0, out1, out2, out3)
plot(out, pch='.')
August 22nd
Assembly of the LuxR generator and the AiiA (BBa_K516022). We cleaved each part with XbaI and PstI (LuxR generator) or EcoRI and SpeI (AiiA). They were run on 1.2% agarose gels and extracted using QIAquick gel extraction kit. Through three way ligation, we managed to clone both inserts into the BBa_J61002 backbone that was precut with with EcoRI and PstI. We confirmed the insertion by amplifying the ligation products with GoTaq polymerase. The shift to 2Kb in lane D indicates the successful ligation reaction. We did transformations with E. coli strain DH5 alpha, and grew them on LB agar supplemented with 100ug/ml ampicilin.

August 23rd
We performed colony PCR to screen for the transformants. After 12 hours of incubation, colonies were small and appeared white, so we left them to grow for three more hours. Most colonies appeared to have the right plasmid as they remained unchanged. 14 white colonies were picked at random and used for the PCR. We observed the same band shift which is positive.

August 24th
We tried to assemble the gold sensing device (BBa_K1127008) with the LuxR/AiiA generators. Unfortunately, it was not successful. We think that the digestion could have been incomplete as bands were observed in the controls.

August 27th
Today, we tried to the assembly again. Instead of the three way ligation, we used the Rapid Assembly of Biobricks (RAB). The parts were amplified with Phusion ® polymerase and digested by single restriction enzyme (SpeI for the BBa_K1127008, and XbaI for the LuxR/AiiA generators). The digests were ligated and subjected to PCR amplification. The band is shifted in lane A that indicates successful ligation.

August 28th
We returned to our favourite cloning techniques - the three way assembly. This time we succeeded. For confirmation, we amplified the ligation products. The shift to about 1.5Kb is shown in the gel photo.

September 9th
For the purpose of characterization, we cloned a reporter gene (GFP or RFP generator) into our construct. Using the three way ligation, we managed to do it successfully. The gel photo shows how we cut off the gels for extraction. We also show the streaked colonies of the transformants. They are green which indicates the presence of GFP (DLig 13E).


Nikola's lab book
His lab book can be found online here.