 "
"
Team:UGent/Results
From 2013.igem.org
| (2 intermediate revisions not shown) | |||
| Line 111: | Line 111: | ||
</p> | </p> | ||
<h1> UV Test </h1> | <h1> UV Test </h1> | ||
| - | <p> Phage transduction was used to perform the deletion of the <i>recA</i> gene after chromosomal evolution. To check whether the gene was successfully knocked out, | + | <p> Phage transduction was used to perform the deletion of the <i>recA</i> gene after chromosomal evolution. To check whether the gene was successfully knocked out, a UV test was developed. The UV light puts a certain amount of stress on the bacterial cells. In respons to this stress, an SOS pathway is triggered in which <i>recA</i> plays an important role. Cells in which the <i>recA</i> gene is deleted will not survive on the UV-treated plate. Colonies in which <i>recA</i> was successfully deleted still can be found on a non-treated backup plate. |
<br><br> | <br><br> | ||
<b>RESULT: </b> | <b>RESULT: </b> | ||
| Line 149: | Line 149: | ||
<li>We subtracted the OD of all the wells with the average OD of the blank and did the same for the fluorescence intensity. This was done to standardize the measurements. </li> | <li>We subtracted the OD of all the wells with the average OD of the blank and did the same for the fluorescence intensity. This was done to standardize the measurements. </li> | ||
<li>Because we used many different plates (and thus some strains were spread over two or more plates), we added a reference strain (strain 8) to all the plates. This strain contains one copy of our construct and thus should give the same F/OD in every well. We used various statistical tests to check if there was a significant difference in average F/OD between the plates of one strain. If these tests resulted in a significant difference of the 8 strain, we could not compare the measurements. Luckily this only happened for a few strains. The extensive execution of these tests can be checked in <a href="https://static.igem.org/mediawiki/2013/d/db/UGent_2013_statistics.pdf" target="_blank">this pdf file</a>.</li></ul><p> | <li>Because we used many different plates (and thus some strains were spread over two or more plates), we added a reference strain (strain 8) to all the plates. This strain contains one copy of our construct and thus should give the same F/OD in every well. We used various statistical tests to check if there was a significant difference in average F/OD between the plates of one strain. If these tests resulted in a significant difference of the 8 strain, we could not compare the measurements. Luckily this only happened for a few strains. The extensive execution of these tests can be checked in <a href="https://static.igem.org/mediawiki/2013/d/db/UGent_2013_statistics.pdf" target="_blank">this pdf file</a>.</li></ul><p> | ||
| - | |||
Thereafter we calculated the F/OD for every well. If the data showed some abnormalities, we tested them for outliers. If the values of the outliers were significantly different than the values of the rest of the data, we excluded these observations and did not use them in our further analysis. | Thereafter we calculated the F/OD for every well. If the data showed some abnormalities, we tested them for outliers. If the values of the outliers were significantly different than the values of the rest of the data, we excluded these observations and did not use them in our further analysis. | ||
<br><br> | <br><br> | ||
| Line 168: | Line 167: | ||
</tr> | </tr> | ||
</table><br> | </table><br> | ||
| - | <h1>CIChE and its | + | <h1>CIChE and its stumbling blocks: our hypothesis</h1> |
<p>For the construction of our plasmid we chose to work with a T7 promoter for the <i>ccdB</i> gene as to avoid leaky expression. Cloning was performed in Top10 cells which do not contain T7 RNA polymerase. Taking into account the troubles we had with the construction of the plasmids with the T7-<i>ccdB</i> insert, we can almost certainly conclude that despite this the cells did experience hindering of the CcdB toxin. Often we obtained no colonies after transformation and if we did, there were mutations in the toxic part of the <i>ccdB</i> gene (see further). We presume that this can only be caused if the <i>ccdB</i> was transcribed by other polymerases who are not specific for the T7 promoter. This probably only occurs at very low frequencies, but as in these strains there is no CcdA present at all even the lowest amount of working CcdB can be lethal for these cells.<br><br> | <p>For the construction of our plasmid we chose to work with a T7 promoter for the <i>ccdB</i> gene as to avoid leaky expression. Cloning was performed in Top10 cells which do not contain T7 RNA polymerase. Taking into account the troubles we had with the construction of the plasmids with the T7-<i>ccdB</i> insert, we can almost certainly conclude that despite this the cells did experience hindering of the CcdB toxin. Often we obtained no colonies after transformation and if we did, there were mutations in the toxic part of the <i>ccdB</i> gene (see further). We presume that this can only be caused if the <i>ccdB</i> was transcribed by other polymerases who are not specific for the T7 promoter. This probably only occurs at very low frequencies, but as in these strains there is no CcdA present at all even the lowest amount of working CcdB can be lethal for these cells.<br><br> | ||
We performed CIChE with the plasmid we constructed ourselves (pSB6A1-T7<i>ccdB</i>) as well as with the original plasmids from which we constructed this part: p5SpFRT-T7<i>ccdB</i>, p10SpFRT-T7<i>ccdB</i> and p20SpFRT-T7-<i>ccdB</i>. According to the GFP measurements we performed, none of these resulted in duplication of the <i>ccdA-gfp</i> construct in the genome. We presume that this is caused by the malfunction of CcdB due to mutations. But how is it possible that there’s so many of these mutations?<br><br> | We performed CIChE with the plasmid we constructed ourselves (pSB6A1-T7<i>ccdB</i>) as well as with the original plasmids from which we constructed this part: p5SpFRT-T7<i>ccdB</i>, p10SpFRT-T7<i>ccdB</i> and p20SpFRT-T7-<i>ccdB</i>. According to the GFP measurements we performed, none of these resulted in duplication of the <i>ccdA-gfp</i> construct in the genome. We presume that this is caused by the malfunction of CcdB due to mutations. But how is it possible that there’s so many of these mutations?<br><br> | ||
Latest revision as of 11:34, 15 October 2013
 |
||||||||||||||||||||||||||||||||||
|
Strains with ccdA-gfp constructThe following strain containing the construct to be duplicated (ccdA-gfp) was constructed (KI experiment 1):
We have two versions of this strain: 5 and 8. The two strains were used in our further experiments as to test possible differences between the two versions. Plasmids containing T7-ccdB
We constructed plasmid pSB6A1-T7ccdB for use in CIChE and pSB1C3-T7ccdB to submit in the biobrick-registry. Through these plasmids, pressure can be put on the CIChE strains simply by adding IPTG. We started with a PCR on p20SpFRT-T7ccdB with primers MDM0586 and MDM0587. We then used restriction enzymes XbaI and PstI to cut the obtained T7-ccdB and the plasmid backbones (pSB6A1 and pSB1C3). After ligating these parts, the DNA was transformed into E. coli Top10 subcloning cells, using heat shock. This experiment delivered pSB6A1 with a deletion of cytosine at position 5929 (for pSB6A1), but failed multiple times for pSB1C3. We then tried to create the plasmid using the Gibson Assembly technique. This was done using T7-ccdB and backbone constructed with specific primers. The result of this transformation (using electroporation) was pSB1C3 with a deletion of adenosine at position 3988 (for pSB1C3). A backmutation was performed on both plasmids using specially constructed primers. This process was succesfull for pSB6A1-T7ccdB, but not for pSB1C3-T7ccdB.
CIChE Strains
The plasmids and strains mentioned above were used in transformations (experiment 3) to obtain strains containing all elements necessary to perform chromosomal evolution. The following strains were constructed:
UV Test Phage transduction was used to perform the deletion of the recA gene after chromosomal evolution. To check whether the gene was successfully knocked out, a UV test was developed. The UV light puts a certain amount of stress on the bacterial cells. In respons to this stress, an SOS pathway is triggered in which recA plays an important role. Cells in which the recA gene is deleted will not survive on the UV-treated plate. Colonies in which recA was successfully deleted still can be found on a non-treated backup plate.
CIChE
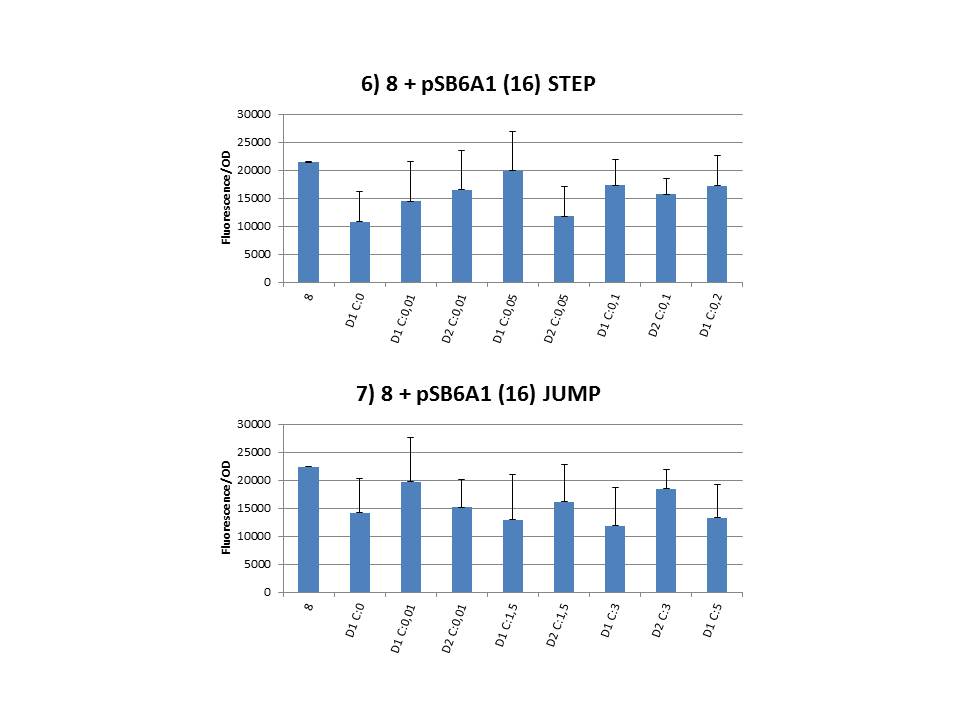
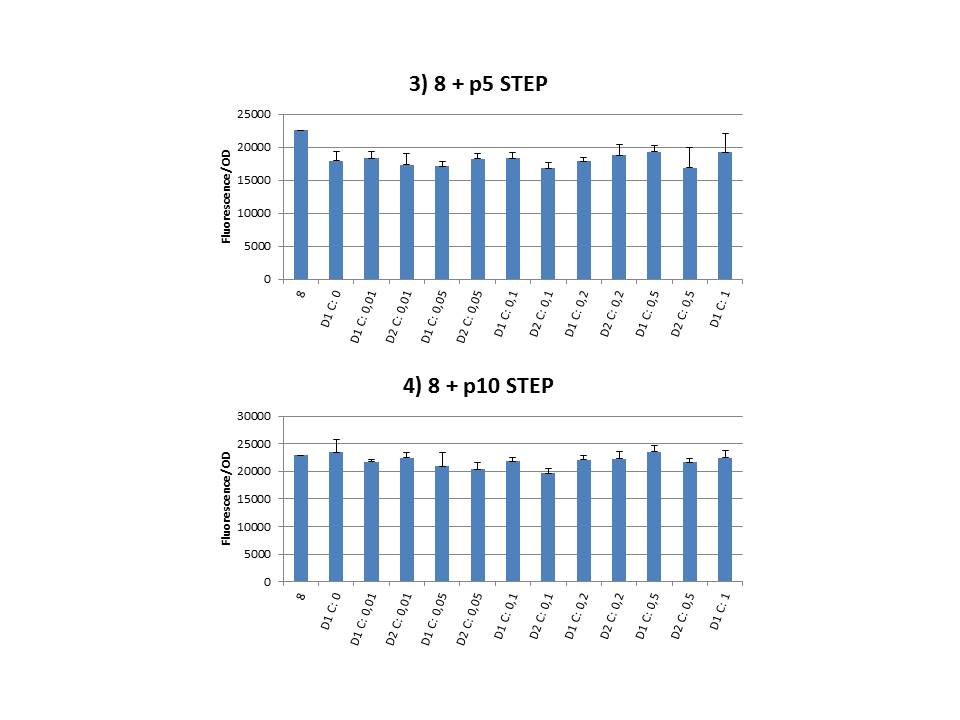
The practical execution of CIChE was as follows: a strain was inoculated with the right concentration of IPTG (day 1 = D1), these strains were grown overnight at 37°C. The next day the strains were inoculated with the same IPTG concentration (day 2 = D2) and with an increased IPTG level (D1). IPTG concentration was raised with a small “step” or with a larger “jump”. These strains were also grown overnight at 37°C. This was repeated over the next days. Because the outcome was difficult to predict, a lot of different IPTG concentrations and ranges were tested.
Thereafter we calculated the F/OD for every well. If the data showed some abnormalities, we tested them for outliers. If the values of the outliers were significantly different than the values of the rest of the data, we excluded these observations and did not use them in our further analysis.
CIChE and its stumbling blocks: our hypothesisFor the construction of our plasmid we chose to work with a T7 promoter for the ccdB gene as to avoid leaky expression. Cloning was performed in Top10 cells which do not contain T7 RNA polymerase. Taking into account the troubles we had with the construction of the plasmids with the T7-ccdB insert, we can almost certainly conclude that despite this the cells did experience hindering of the CcdB toxin. Often we obtained no colonies after transformation and if we did, there were mutations in the toxic part of the ccdB gene (see further). We presume that this can only be caused if the ccdB was transcribed by other polymerases who are not specific for the T7 promoter. This probably only occurs at very low frequencies, but as in these strains there is no CcdA present at all even the lowest amount of working CcdB can be lethal for these cells. Clone Manager and I-TASSERIn order to examine the effect of these mutations on the protein structure, we determined the amino acid sequence of the mutated plasmid 8 + pSB6A1-T7ccdB, 2 mM IPTG day 2, Step using Clone Manager:
MAQFKVYTYKRESRYRLFVDVQSDIIDTPGRRMVIPLASARLLSDKVSRELYPVVHIGDESWRMMTTDMASVPVSVI
At the position of the Tryptophan residue responsible for interaction with Arg462 and consequently essential for toxic activity, there is now a Glycine residue. Also the following two amino acids essential for toxicity are mutated. Furthermore chances are that the following part of the protein will sterically hinder interaction with DNA gyrase. As a conclusion this amino acid sequence is a good indication that this mutated CcdB protein won’t be able to execute its toxic function.
To further investigate the consequences of the frameshift we used I-TASSER to predict the structure of this protein, resulting in following top 5 of predicted models:
Note: C-score is typically in the range of [-5,2], where a C-score of higher value signifies a model with a high confidence and vice-versa.  Further research perspectives
Assemble T7-ccdB plasmid inside CcdB-survival strains SourcesDao-Thi, M.H. et al. Molecular basis of gyrase poisoning by the addiction toxin CcdB. Journal of Molecular Biology 348, 1091-1102 (2005).Couturier, M., Bahassi, E. & Van Melderen, L. Bacterial death by DNA gyrase poisoning. Trends in Microbiology 6, 269-275 (1998). Corzett, C.H., Goodman, M.F. & Finkel, S.E. Competitive Fitness During Feast and Famine: How SOS DNA Polymerases Influence Physiology and Evolution in Escherichia coli. Genetics 194, 409-420 (2013).
|

Tweets van @iGEM_UGent |
|||||||||||||||||||||||||||||||||
















|
|




