Notebook
Transformed BioBricks
Tuesday 18.06.13
Started transforming BioBricks. Transformed some composite BioBricks consisting of constitutive promotor, RBS, a fluorescent protein and a terminator. Transformed the parts Promoter+RBS+RFP+term (<partinfo>BBa_I13521</partinfo>), Promoter+RBS+GFP+term (<partinfo>BBa_I13522</partinfo>), Promoter+RBS+CFP+term (<partinfo>BBa_I13600</partinfo>) and Pm+RBS+mCherry+term to competent E.coli DH5α cells, to have some colorful cells to show the camera crew from NRK (Norwegian broadcasting), who will visit our lab tomorrow :-)
For our transformation protocol, see the protocol page.
Wednesday 19.06.13
We transformed BioBricks consisting of fluorescent proteins. Transformed the parts YFP (<partinfo>BBa_E0030</partinfo>), CFP (<partinfo>BBa_E0020</partinfo>), RFP (<partinfo>BBa_E1010</partinfo>), GFP (<partinfo>BBa_E0040</partinfo>), BFP (<partinfo>BBa_K592100</partinfo>) and SYFP (<partinfo>BBa_K864100</partinfo>).
We transferred mCherry from agar plate to liquid medium and incubated at 37° C.
Thursday 20.06.13
We transferred the transformed cells from yesterday to liquid medium and incubated at 37°C protocol.
We isolated the DNA from the transformed mCherry cells by using the miniprep protocol and measured the concentration by [http://www.nanodrop.com/library/nd-1000-v3.7-users-manual-8.5x11.pdf NanoDrop ND-1000 Spectrophotometer].
| Sample
| Concentration (ng/µl)
|
| mCherry1
| 5,6
|
| mCherry2
| 7,3
|
Friday 21.06.13
We isolated the DNA from the transformed cells by using the miniprep protocol and measured the concentration by [http://www.nanodrop.com/library/nd-1000-v3.7-users-manual-8.5x11.pdf NanoDrop ND-1000 Spectrophotometer].
| Sample
| Concentration (ng/µl)
|
| CFP1
| 8,59
|
| CFP2
| 7,04
|
| BFP
| 7,06
|
| SYFP
| 5,42
|
| RFP1
| 5,04
|
| RFP2
| 7,52
|
| YFP
| 6,79
|
| GFP
| 5,27
|
Monday 24.06.13 - Friday 28.06.13
This week we have established protocols for further work and designed primers for PCR.
Monday 01.07.13
We transformed E.coli strain DH5α with some BioBricks: pBAD promoter (<partinfo>BBa_K206000</partinfo>), RBS (<partinfo>BBa_J61101</partinfo>) and a plasmid backbone (<partinfo>BBa_J01101</partinfo>).
Tuesday 02.07.2013
Small-scale vesicle preparation
This day we started making the small-scale vesicle preparation according to our protocol.
Overnight cell cultures of E.coli ER2566 and E.coli BW27784 transformed with the pUM9 plasmid were preprared. The pUM9 plasmid has ampR induces a stress respons in E.coli when arabinose is present. According to the litterature (link?) stress in E.coli increases vesicle production.
The ER2566 cell were cultured in plain LB, while the BW27784 cells were cultured in LB with ampenicillin (100 µg/mL).
Transfer to liquid medium
We transferred the samples of tranformed DH5α (pBAD promoter (<partinfo>BBa_K206000</partinfo>), RBS (<partinfo>BBa_J61101</partinfo>) and a plasmid backbone (<partinfo>BBa_J01101</partinfo>)) from agar plate to liquid medium and incubated at 37°C overnight.
Wednesday 03.07.2013
Small-scale vesicle preparation
The small-scale vesicle preparation continued with completion of step 2-7 in the vesicle isolation protocol. There where made three cell cultures in step 2; E.coli ER2566 in LB, E.coli BW27784 in LB with ampenicillin (100 µg/mL) (ara-) and E.coli BW27784 in LB with ampenicillin (100 µg/mL) with the addition of arabinose (0.5 %) after 1 hour of incubtion (ara+). The cell cultures in step 2 was incubated for 3 hours.
Optical denisty (OD) at 600 nm was mesured as indicated in the table below. No dilution of the samples was necessary.
| Sample
| OD600
|
| ER2566
| 0.9805
|
| ara-
| 0.1263
|
| ara+
| 0.0471
|
DNA isolaton from transformants
We isolated the DNA from the transformed cells (pBAD promoter (<partinfo>BBa_K206000</partinfo>), RBS (<partinfo>BBa_J61101</partinfo>) and a plasmid backbone (<partinfo>BBa_J01101</partinfo>)) by using the miniprep protocol and measured the concentration by [http://www.nanodrop.com/library/nd-1000-v3.7-users-manual-8.5x11.pdf NanoDrop ND-1000 Spectrophotometer].
| Sample
| Concentration (ng/µl)
|
| RBS
| 4,91
|
| pTet1
| 8,99
|
| pTet2
| 4,39
|
| pBAD1
| 3,54
|
| pBAD2
| 9,24
|
Thursday 04.07.2013
Continuation of the small-scale vesicle preparation according to the vesicle isolation protocol. Step 8-12 was completed with the exception that the pellet was resuspended in 0.5 mL DPBSS insted of 100 µL in step 11 and that the check for sterility in step 12 was not performed. The SDS-PAGE showed no signs of vesicles as viewed in figure 1.
 figure 1
figure 1
Monday 08.07.10
An overnight culture was started for vesicle isolation as desribed for tuesday 02.07.10.
Tuesday 09.07.2013
Today we ran PCR on 4 FP's <partinfo>Bba_K864100</partinfo>, <partinfo>Bba_K592100</partinfo>, <partinfo>Bba_E0040</partinfo>, <partinfo>Bba_E1010</partinfo> and 1 backbone <partinfo>Bba_J01101</partinfo>. We used the Phusion DNA Polymerase protocol from New England Biolabs [1] for 50 ul sample(without DMSO). All amplicons were run on a GelGreen 0.8% agarose gel. We only got a band on our RFP <partinfo>Bba_E1010</partinfo>, and it was not the expected size. We will adjust the PCR parameters and run them again tomorrow.
Step 2-11 of the small scale-vesicle preparation protocol and step 1-4 of the density gradient protocol was performed. Samples from the small-scale vesicle preparation were made ready for SDS-PAGE.
The incubation period was revised from the first attempt to isolate vesicles. The duration and other condition is summerised in the table below.
|
| Sample
| Sample
| Sample
|
| Time (h:min)
| ER2566
| MW27784 ara-
| MW27784 ara+
|
| 0:00
|
| Add 250 μL Amp (100X) and 2,5 mL of overnight culture to 250 mL of LB. Incubate at 37 C
| Add 250 μL Amp (100X) and 2,5 mL of overnight culture to 250 mL of LB. Incubate at 37 C
|
| 1:00
| Add 1 mL of overnight culture to 250 mL of LB. Incubate at 37 C
|
|
|
| 2:00
|
|
| Add 1,25 μL arabinose (1000X)
|
| 4:00
| Measure OD-600
| Measure OD-600
| Measure OD-600
|
Optical denisty (OD) at 600 nm was mesured in step 4 as indicated in the table below. No dilution of the samples was necessary.
| Sample
| OD600
|
| ER2566
| 0.1671
|
| ara-
| 0.6726
|
| ara+
| 0.5968
|
Wednesday 10.07.2013
We ran the PCR again with different parameters. Increased annealing- and elongationtime and a separate program for the backbone due to its larger size. We still only got a band for RFP <partinfo>Bba_E1010</partinfo>. In the prosess of rechecking the primers it was discovered that the primers for RFP are incorrect which explains the discrepancy in productsize. The other primers should be working so tomorrow we will redo the dilutions of the primers, increase the elongation time and lower the annealing temperature a little, add DMSO, add a bit larger templatesample and use 1-step PCR.
SDS-PAGE were run with the samples from the vesicle preparaton from the day before. There were no indication of vesicles in the samples as indicated on the gel (figure 2)
 figure 2
figure 2
Step 5 of the purification of vesicles by density gradient was performed. The fraction were freezed down at -80 oC.
Thursday 11.07.2013
Today we ran PCR again. This time we omitted the RFP, since we discovered that one of the primers was incorrect. We diluted the primers one more time and changed the parameters for the PCR. This time we only did one step, with 30 cycles. We also decreased the annealing temperature and increased the elongationtime. The results were more uplifting this time around after running gel electrophoreses (picture coming soon). We got a clear band on the backbone and faint bands on the GFP and SYFP. Since there was a faint band on the backbone of a larger size than the one we wanted, we added 1 µl of Dpn1 to the amplicon and put it in a 37 °C waterbath for an hour. We used the QIAGEN PCR purification kit to isolate the DNA and measured the cioncentration by [http://www.nanodrop.com/library/nd-1000-v3.7-users-manual-8.5x11.pdf NanoDrop ND-1000 Spectrophotometer].
| Sample
| Concentration (ng/µl)
|
| SYFP
| 13,16
|
| GFP
| 55,62
|
| BB
| 55,50
|
Friday 12.07.2013
We repeated the PCR on the SYFP and BFP. We changed some of the conditions (increased number of cycles and varying the template volume), but still no result.
Sunday 14.07.2013
Step 1 and 2 of the Small-scale vesicle preparation were performed. 5 mL cultures of the E.coli strains ER2566, DH5α and MW27784 were incubated in 5 mL LB at 37 oC for 8 h. 2,5 mL of these cell cultures were added to 250 mL of LB and incubated at 37 oC for 13 h.
Monday 15.07.2013
Small scale-vesicle preparation protocol
Step 3-11 of small scale-vesicle preparation protocol and step 1-4 of the density gradient protocol was performed. Samples from the small-scale vesicle preparation were made ready for SDS-PAGE.
Optical denisty (OD) at 600 nm was mesured in step 4 as indicated in the table below. The samples were diluted 1:16 with LB media.
| Sample
| OD600
|
| ER2566
| 0.133
|
| DH5α
| 0.169
|
| MW27784
| 0.198
|
A relative concentration of vesicles from the small scale preparation was mesured by adding the hydrophobic fluorecent dye [http://products.invitrogen.com/ivgn/product/T3166 FM4-64] and measuring fluorecence (RFU) (as desribed in step 12 in the small-scale vesicle preparation). Results (table below) indicated presence of vesicles in the ER2566-sample only.
| Sample
| RFU (exitation/emission at 506/750 nm)
|
| Empty
| 3
|
| Blank
| 19 814
|
| MW27784
| 19 598
|
| DH5α
| 19 254
|
| ER2566
| 40 987
|
New rounds of PCR
Two rounds of PCR was run today with varying the conditions.There are still no result. For the BFP, we will wait for new primers, since the result are showing formation of primer dimers. A new transformation of the SYFP-biobrick was started to get a new template for the PCRs.
Tuesday 16.07.13
SDS-PAGE were run with the samples from the vesicle preparaton from the day before. The ER2566-sample had vesicles, where as the other samples did not contain any (figure 3)
 figure 3: Ladder applied is Precision Plus ProteinTM Unstained Standards.
figure 3: Ladder applied is Precision Plus ProteinTM Unstained Standards.
Step 5 of the purification of vesicles by density gradient was performed. SDS-PAGE were run on the fractions from ER2566, as this was the only sample with any indication of vesicles. As indicated in figure 4, fraction 4 and 5 had clear bands, whereas the fraction 6 has pale bands. The fractions are numbered according to when whey where removed from the gradient with fraction 1 beeing the first fraction to be removed and so on.
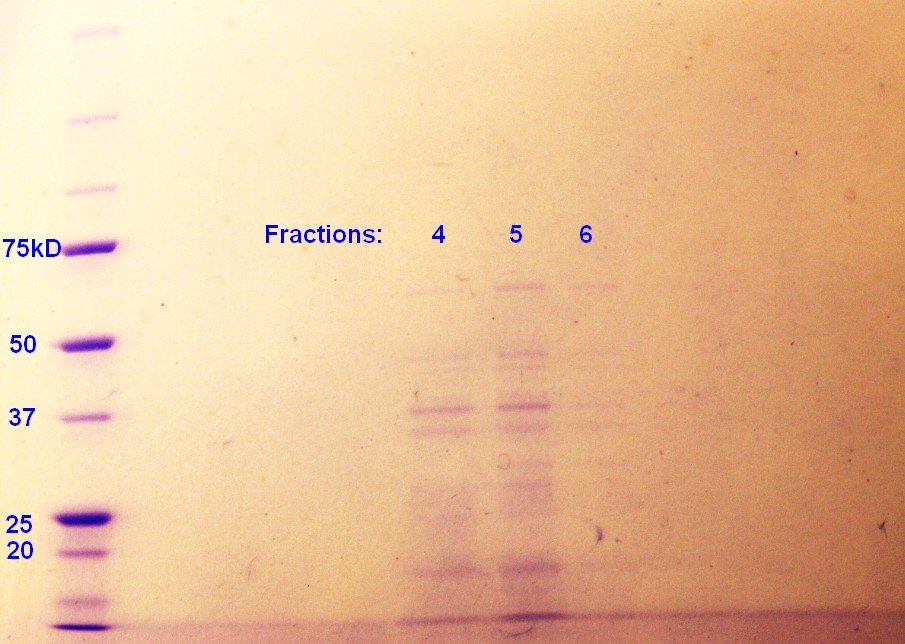 figure 4: Ladder applied is Precision Plus ProteinTM Unstained Standards
figure 4: Ladder applied is Precision Plus ProteinTM Unstained Standards
The rest of the fractions were freezed down at -80 oC. Too short incubation time in step 2 of the small-scale vesicle preparation seem to be the reason why vesicles were not isolated during the two previous attemts.
Due to postive results, the Small-Scale vesicle preparation was repeated: Step 1 and 2 of the Small-Scale vesicle preparation was performed. 5 mL cultures of the E.coli strains ER2566, DH5α and MW27784 were incubated in 5 mL LB at 37 oC for 8 h. 2.5 mL of these cell cultures were added to 250 mL of LB and incubated at 37 oC for 14 h.
Wednesday 17.07.13
Small-Scale vesicle preparation
Step 3-11 of the Small-Scale vesicle preparation protocol was performed on the ER2566, DH5α, and MW27784-samples.
Optical denisty (OD) at 600 nm was mesured in step 4 as indicated in the table below. The samples were diluted 1:10 with LB media.
| Sample
| OD600
|
| ER2566
| 0.281
|
| DH5α
| 0.293
|
| MW27784
| 0.402
|
The DH5α had no pallet after the second centrifugation, and this sample where therefore not included further in the experiement. Some of MW27784 sample was lost in step 10 of the small-scale vesicle preparation. A part of the ER2566 and MW27784-samples from the small-scale vesicle preparation were made ready for SDS-PAGE. The rest of the samples were mixed with OptiPrep (60%) to give a final OptiPrep concentration of 45% and frozen at -80oC.
A relative concentration of vesicles from the small scale preparation was mesured by adding the hydrophobic fluorecent dye [http://products.invitrogen.com/ivgn/product/T3166 FM4-64] and measuring RFU (as desribed in step 12 in the small-scale vesicle preparation). This time we also measured RFU with exitation at 515 nm and emission at 640 nm. As the last exitation and emission gave a more significant difference between the blank sample and the vesicle containing sample, this condition will be applied for later RFU-measurements. Results (table below) indicated presence of vesicles in the ER2566-sample and perhaps also in the MW27784-sample.
| Sample
| RFU (exitation/emission at 506/750 nm)
| RFU (exitation/emission at 515/640 nm)
|
| Empty
| 1
| 32
|
| Blank
| 16 536
| 1 180
|
| MW27784
| 16 335
| 1 691
|
| ER2566
| 47 191
| 44 059
|
DNA isolation
We isolated the DNA from the transformed cells (with SYFP) by using the miniprep protocol and measured the concentration by [http://www.nanodrop.com/library/nd-1000-v3.7-users-manual-8.5x11.pdf NanoDrop ND-1000 Spectrophotometer].
| Sample
| Concentration (ng/µl)
|
| SYFP1
| 30,5
|
| SYFP2
| 20,8
|
We used the new isolated DNA as template in PCR, trying different parameters.
Thursday 18.07.13
SDS-PAGE were run with the samples from the vesicle preparaton from the day before. The ER2566-sample had a clear content of vesicles whereas the MW27784-sample had a less denser content of vesicles (figure 5). The MW27784 also seems to have a different protein profile.
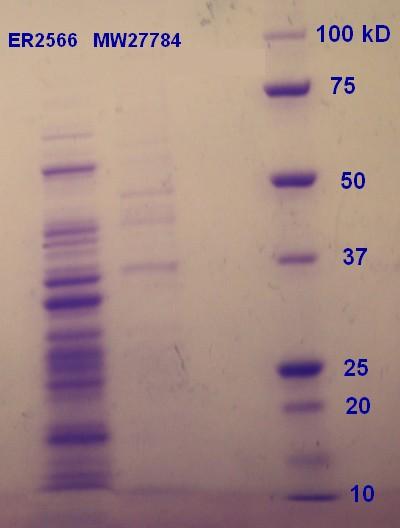 Figure 5: Ladder applied is Precision Plus ProteinTM Unstained Standards.
Figure 5: Ladder applied is Precision Plus ProteinTM Unstained Standards.
Friday 19.07.13
E.coli strain ER2566 was transformed with biobrick [http://parts.igem.org/Part:BBa_K530015 BBa_K530015] according to the transformation protocol. The plates were incubated for 19 hours and then moved to the fridge. The transformed ER2566 bacteria is later to be used for determination of influence of chloramphenicol on the cells and for growth duration for optimal vesicle production.
Monday 22.07.13
Three cell cultures of E.coli strain ER2566 transformed with [http://parts.igem.org/Part:BBa_K530015 BBa_K530015] was inoculated in Chl-LB and one cell culture of MW27784 were inoculated in LB for 7.5 hours at 37°C as described in step 1 of the small-scale vesicle preparation. The transformed ER2566 seems to grow slower in Chl_LB than untransformed ER2566 does in plain LB. Bigger overnight cell cultures were incubated as in step 2 of the small-scale vesicle preparation, the ER2566 cells were incolated in Chl-LB as before. The three ER2566 samples were incubated for 14, 16 and 18 hours, while the MW27784 sample was incubated for 16 hours.
Tuesday 23.07.13
Step 4-12 of the small-scale vesicle preparation were performed. In addition dilutions (10-2, 10-4 and 10-6) of the ER2566 samples were made and plating of 100µL of 1 mL of the dilutions on Chl-LA was performed in step 4. The plates were left in the incubator overnight.
Optical denisty (OD) at 600 nm was mesured in step 4 as indicated in the table below. The samples were diluted 1:10 with LB media.
| Sample
| OD600
|
| ER2566 14h
| 0.247
|
| ER2566 16h
| 0.347
|
| MW27784 16h
| 0.533
|
| ER2566 18h
| 0.328
|
Samples for SDS-PAGE was prepared in step 12.
RFU was measured as decribed in step 12 of the small-scale vesicle preparation protcol. Results were as indicated in the table below:
| Sample
| RFU (exitation/emission at 515/640 nm)
|
| Empty
| 91
|
| Blank
| 6 745
|
| ER2566 14h
| 31 571
|
| ER2566 16h
| 45 089
|
| MW27784 16h
| 6 899
|
| ER2655 18h
| 22 328
|
Wednesday 24.07.13
Small-scale vesicles isolation
Plates with transformed ER2566, plated the day before, were checked for colonies. As the 10-2 and 10-4 dilution had to much growth, only the 10-6-dilutions were counted for colony forming units (CFU)(table below):
| Sample
| CFU (10-6-dilution)
| CFU/mL (undiluted)
|
| ER2566 14h
| 265
| 2.65*109
|
| ER2566 16h
| 278
| 2.78*109
|
| ER2566 18h
| 273
| 2.73*109
|
It seems that transformed ER2566 reaches steady state at 14h or quickly after 14h in the 240 mL cellmedia. The CFU results corresponds well with the measured OD600 (table from the day before) except for that the OD600 of ER2566 14h is more different from the other samples compared to the CFU-results.
SDS-PAGE were run on the samples prepared the day before. There were very weak bands on the on all of the ER2566 samples (not as visible in the photograph (figure 6) as in real life) and no indication of vesicles on the MW27784-sample.
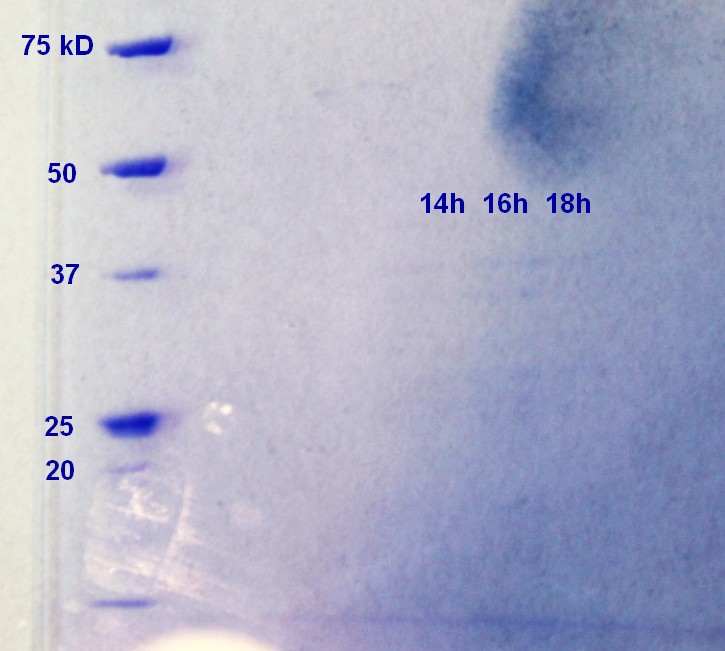 Figure 6: Ladder applied is Precision Plus ProteinTM Unstained Standards.
Figure 6: Ladder applied is Precision Plus ProteinTM Unstained Standards.
The results of the SDS-PAGE does not correspond well to the results of the RFU-measurements from the day before. The RFU results indicate that the concentration of vesicles is higher.
New Primers
We recieved the new primers for the GFP, RFP and BB(Backbone) that we intend to attach to each other with Gibson Assembly. We got results from all and purified DNA using NEB Purification Kit.
Tranformation
Since we cannot seem to get the primers for SYFP and BFP to work and they are both developed by the same iGEM team we're testing them for futher characterization. Transformation of SYFP, BFP and an appropritate BB with promoter and RBS were done and plated out. The plates are incubated in 37 degrees overnight and put in the frige.
Thursday 25.07.13
Because the SDS-PAGE from yesterday did not give us expected results in terms of vesicles consentration, we started with the density gradient vesicle isolation with the ER2566 14h and 16h-samples to further test them for vesicles. Step 1-4 of this protocol was performed.
Isolation of Tat-sequence
We isolated the tat-signaling sequence beloning to the TorA gene from the "E. Coli" strain ER2566 using our designed primers with PCR.
Friday 26.07.13
Density gradient for vesicle purification
Step 5 and 6 of the density gradient vesicle isolation protocol was performed. The SDS-PAGE of the fractions show no or little indication of vesicles, further confirming the tendencies from the day before.
Gibson assembly
The Tat-PCR product was purified using NEB DNA Purification Kit. We will use our purified PCR products of Tat, GFP, RFP and BB in Gibson assembly.
The Gibson assembly reaction of the plasmid backbone, tat signal-sequence, GFP and RFP to create our first construct was performed according to the Gibson AssemblyTM Cloning Kit Manual. We made two samples, one with a incubation time (with the mastermix) of 60 min and one with 15 min. The control-sample was incubated for 15 min. The samples were then used for transformation of both E.coli strain DH5α and ER2566, giving us 4 samples. The transformation protocol was also collected from the Gibson AssemblyTM Cloning Kit Manual. Transformed bacteria were incubated on plates with the appropriate antibiotics for 18 hours and then put in the fridge.
| Insert(I)/Vector(V)
| Concentration(ng/µl)
| Size(bp)
| µl
|
| Tat(I)5X
| 35.84
| 150
| 1.5
|
| GFP(I)2X
| 52.68
| 750
| 1.5
|
| RFP(I)2X
| 98.31
| 750
| 1.0
|
| BB(V)
| 73.82
| 2500
| 1.4
|
Add 10 µl of Gibson Mastermix and up to 20 µl dH2O.
There were no colonies on the plates where bacteria transformed with the 60 minutes incubation time Gibson assembly plasmids. There were three colonies with ER2566 transformed with the 15 minutes incubation time Gibson assenmbly plasmids and one colony on the plate with DH5α.
Sunday 28.07.13
The colonies with SYFP, BFP and BB was transferred to liquid media.
Monday 29.07.13
Small-scale vesicle preparation
Due to our unexpected vesicle yeild last week (see figure 6), we started a new round of small-scale vesicle preparation. Step 1 and 2 of this protocol was performed with E.coli strains DH5α, untransformed ER2566 and ER2566 transformed with biobrick [http://parts.igem.org/Part:BBa_K530015 BBa_K530015]. The transformed ER2566 was inculated in LB with chloramphenicole. All cultures were incubated for 16 hours.
Miniprep
Isolation of SYFP, BFP and BB from last weeks transformation.
Restriction Digest
The GFP, BFP an BB wad digested using restrictionenzymes. We followed the protocol [http://parts.igem.org/Help:Protocols/Restriction_Digest Restricion Digest]. BB was digested with SpeI and PstI and SYFP/BFP with XbaI and PstI. The following ligation was performed using this [http://parts.igem.org/Help:Protocols/Ligation Ligation Protocol] using bot 1:1 and 1:2 vector insert ratio.
Tuesday 30.07.13
Small-Scale vesicle preparation
Step 4-12 of the small-scale vesicle preparation were performed. In addition a 10-6 dilution of the ER2566 samples were made and plating of 100µL of 1 mL of the dilution on Chl-LA was performed in step 4. The plates were left in the incubator overnight. The plate had growth, indicating that the bacteria still had their plasmid.
Optical denisty (OD) at 600 nm was mesured in step 4 as indicated in the table below. The samples were diluted 1:10 with LB media.
| Sample
| OD600
|
| Unstransformed ER2566
|
|
| Transformed ER2566
|
|
| Unstransformed DH5α
|
|
Samples for SDS-PAGE was prepared in step 12.
RFU was measured as decribed in step 12 of the small-scale vesicle preparation protcol. Results were as indicated in the table below:
| Sample
| RFU (exitation/emission at 515/640 nm)
|
| Empty
| 93
|
| Blank
| 22340
|
| Transformed ER2566
| 27249
|
| Unstransformed ER2566
| 48478
|
| Unstransformed DH5α
| 22029
|
From now on we will consentrate on E.coli strain ER2566.
Analysis of Gibson Assembly construct
Miniprep of Gibson Assembly construct and restriction digest gave wrong product on the gel. The size is not correct, so we must try again.
Check for contamination
To ensure that there is no contamination in our 4 construct component DNA we put 1µl of each DNA sample in 50µl of competent cells(ER2566) and plated them out. Incubated at 37°C overnight.
Second Gibson Assembly
We used the same protocol, but changed the DNA concentration to 200 ng and increased the ratio of tat to 10X instead of 5X.
| Insert(I)/Vector(V)
| Concentration(ng/µl)
| Size(bp)
| µl
|
| Tat(I)10X
| 35.84
| 150
| 2.5
|
| GFP(I)2X
| 52.68
| 750
| 3.0
|
| RFP(I)2X
| 98.31
| 750
| 1.5
|
| BB(V)
| 73.82
| 2500
| 3.0
|
Add 10 µl Gibson Mastermix.
They where then transformed into ER2566 cells on 4 plates. Three plates with 5µl template, and one with standard 2µl template. Incubated at 37°C overnight.
| Plate
| Templatevolume(µl)
| Colonies
|
| 1
| 2
| 3
|
| 2
| 5
| 36
|
| 3
| 5
| 37
|
| 4
| 5
| 250
|
Colonies where picked from all four plates and transferred to liquid media. Left overnight in 37 degrees shaker.
Wednesday 31.07.13
Small-scale vesicle preparation
SDS-PAGE were run on the samples from the day before. There were only a few weak bands on the gel (not visible on a potential picture) indicating vesicles in the unstransformed ER2566-sample only.
A new round of small-scale vesicle preparation was started with two transformed ER2566 samples (same conditions) and one sample of unstransformed ER2566. The transformed ER2566 had a plasmid that wes a product from the first Gibson Assenbly. Step 1 and 2 of the protocol was completed with the transformed ER2566 incubating in LB with chloramphenicol (25µL/mL) and untransformed ER2566 incubating in plain LB.
Restriction Digest of Gibson Assembly
We followed the procedure [http://parts.igem.org/Help:Protocols/Restriction_Digest Restriction Digest] in the registry.
| Template
| Concentration(ng/µl)
| µl
| dH2O
|
| 1
| 17.9
| 14
| 2
|
| 2
| 137
| 2
| 14
|
| 3
| 26.7
| 9.5
| 6.5
|
| 4
| 20.6
| 12
| 4
|
Incubated at 50 °ree;C for 15 min. Run on gel, positive result for sample 1. Will do confirmation PCR tomorrow.
Thursday 01.08.13
Small-scale vesicle preparation
Step 4-7 of the small-scale vesicle preparation were performed. In addition a 10-6 dilution of the ER2566 samples were made and plating of 100µL of 1 mL of the dilution on Chl-LA was performed in step 4. The plates were left in the incubator overnight. The plate had growth, indicating that the bacteria still had their plasmid.
Optical denisty (OD) at 600 nm was mesured in step 4 as indicated in the table below. The samples were diluted 1:10 with LB media.
| Sample
| OD600
|
| Transformed ER2566 (1)
| 0.34
|
| Transformed ER2566 (2)
| 0.32
|
| Unstransformed ER2566
| 0.37
|
Due to complications in step 7, this vesicle isolation attempt was terminated.
Confirmation PCR of Gibson Construct
To confirm that our result from yesterday was in fact our construct with tat-sequense, GFP and RFP we performed several PCR reactions with different targets. We used 5 templates, 2 samples from the 2µl plats and 3 from the 5µl plates. All run with 4 different primersets. Tm was calculated using Tm Caculator. Only the actual primer, not the flankingsequence was included in the calculations. The Phusion PCR Protocol with 20µl sample was used.
| Target
| F_Primer
| R_Primer
| Tm
|
| GFP_RFP
| F_tat_GFP
| R_RFP
| 58°C
|
| Tat_GFP_RFP
| F_pl.b_tat
| R_RFP
| 64/72°C
|
| Tat_GFP
| F_pl.b_tat
| R_GFP
| 58°C
|
| Tat
| F_pl.b_tat
| R_tat
| 72°C
|
All were run on GelGreen 0.8% Agarose. Bands indicate no GFP, tat or tat_GFP_RFP construct present, and tat_RFP-construct(which should not be possible) present.
|
Friday 02.08.13
Transformation
Biobrick [http://parts.igem.org/Part:BBa_J04450 BBa_J04450] (in plasmid with kanamycin resistance) was transformed into E.coli strain ER2566 according to our transformation protocole.
Saturday 03.08.13
Step 1 and 2 of the were performed small-scale vesicle preparation with E.coli strains ER2566. Untransformed ER2566 was incubated for 14 hours, transformed ER2566 (with biobrick [http://parts.igem.org/Part:BBa_J04450 BBa_J04450]) for 14 and 16 hours in step 2. The untransformed sample was incubated in plain LB whereas the transformed samples were incubated in LB with kanamycin (50µL/mL)
Sunday 04.08.13
Step 4-12 of the small-scale vesicle preparation were performed. In addition a 10-6 dilution of the transformed ER2566 samples (both 14 and 16 hour incubation time) were made and plating of 100µL of 1 mL of the dilution on Kan-LA was performed in step 4. The plates were left in the incubator overnight. The plates had growth, indicating that the bacteria still had their plasmid.
Optical denisty (OD) at 600 nm was mesured in step 4 as indicated in the table below. The samples were diluted 1:10 with LB media.
| Sample
| OD600
|
| Unstransformed ER2566 14h
| 0.324
|
| Transformed ER2566 14h
| 0.449
|
| Unstransformed ER2566 16h
| 0.493
|
Samples for SDS-PAGE was prepared in step 12.
RFU was measured as decribed in step 12 of the small-scale vesicle preparation protcol. Results were as indicated in the table below:
| Sample
| RFU (exitation/emission at 515/640 nm)
|
| Empty
| 100
|
| Blank
| 4 656
|
| Untransformed ER2566 14h
| 24 549
|
| Transformed ER2566 14h
| 45 732
|
| Transformed ER2566 16h
| 30 655
|
Another round of Small-scale vesicle preparation
Step 1 and 2 of the small-scale vesicle preparation were performed with untransformed ER2566 that was incubated for 14 hours in plain LB in step 2, transformed ER2566 ( with biobrick [http://parts.igem.org/Part:BBa_K530015 BBa_K530015]) that were incubated in LB with chloramphenicole (25 µL/mL) for 16 hours and the same transformed ER2566 in plain LB for 14 hours (incubated WITH chloramphenicole (25 µL/mL) in step 1).
Monday 04.08.13
Small-scale vesicle preparation
Step 4-6 of the small-scale vesicle preparation were performed. In addition a 10-6 dilution of the transformed ER2566 samples (both 14 and 16 hour incubation time) were made and plating of 100µL of 1 mL of the dilution on Kan-LA was performed in step 4. The plates were left in the incubator overnight. The plates had growth, indicating that the bacteria still had their plasmid. This result also means that transformed ER2566 with Chl-resistance can hold on to their plasmid even without a selective marker in the media for at least 14 hours of incubation.
Optical denisty (OD) at 600 nm was mesured in step 4 as indicated in the table below. The samples were diluted 1:10 with LB media.
| Sample
| OD600
|
| Unstransformed ER2566 14h
| 0.379
|
| Transformed ER2566 14h -Chl
| 0.369
|
| Unstransformed ER2566 16h +Chl
| 0.373
|
The experiment was terminated after step 6 because we will not be needing the results.
SDS-PAGE of vesicle samples
SDS-PAGE was run on the samples from the day before. As figure 7 shows the unstransformed sample and the transformed ER2566 14h had a clear indication of vesicles. According to the SDS-PAGE the untransformed sample had a greater content of protein compared to ransformed ER2566 14h, whereas the transformed samples had more vesicles than the unstransformed sample judging from the RFU measurements (table, sunday 04.08.13). It seems that transformation with antibiotics in the media increases vesicle production, but decreases production of vesicle assosiated proteins.
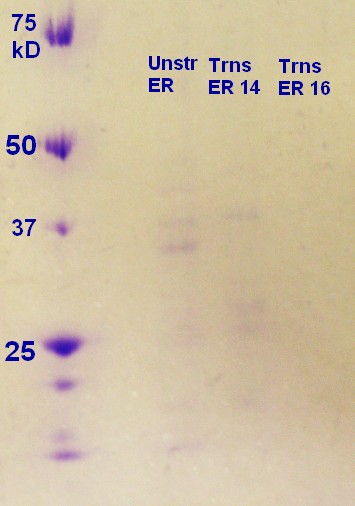 Figure 7: Ladder applied is Precision Plus ProteinTM Unstained Standards.
Figure 7: Ladder applied is Precision Plus ProteinTM Unstained Standards.
Yet another round of small-scale vesicle preparation
Step 1 and 2 of the small-scale vesicle preparation were performed with ER2566 transformed with construct from the second Gibson assembly attempt. Two colonies (see picture 8) were selected and incubated for 14 and 16 hours in step 2. All of the cultures were incubated in LB media with ampenicillin (100 µL/mL).
figure 8
Different PCR with Gibson construct
The results from the last confirmation PCR gave strange results, so it was decided to run it again. Changes where; the whole primer including the flanking sequence to calculate the annealing temperature, included the RFP only, and we used 2-step PCR. The results were the same. Confirmed RFP and tat_RFP, but no tat_GFP_RFP construct. It is likely that the GFP is not present in our product. A new transformation was performed with the GFP to use in next Gibson Assembly.
The tat_RFP construct was purified and frozen at -20°C.
Tuesday 06.08.13
Small-scale vesicle preparation
Step 4-12 of the small-scale vesicle preparation were performed. In addition a 10-6 dilution of all the samples were made and plating of 100µL of 1 mL of the dilution on Amp-LA was performed in step 4. The plates were left in the incubator overnight. The plates had growth, indicating that the bacteria still had their plasmid.
Optical denisty (OD) at 600 nm was mesured in step 4 as indicated in the table below. The samples were diluted 1:10 with LB media.
| Sample
| OD600
|
| 14-1
| 0.432
|
| 14-2
| 0.391
|
| 16-1
| 0.425
|
| 16-2
| 0.420
|
The cell cultures and cell pallets were not red or pink when they were taken out of the incubator and centrifuge. The pallet did, however, start to get a red color after a while. This indicate that the cells reqiure a more aerobic enviroment for production of RFP.
Samples for SDS-PAGE was prepared in step 12.
RFU was measured as decribed in step 12 of the small-scale vesicle preparation protcol. As the vesicles were expected to contain RFP, RFU was also measured without FM64-4 at exitation/emission at 605/670 (the known exitation/emission for RFP) Results were as indicated in the table below:
| Sample
| RFU (exitation/emission at
515/640 nm) with FM64-4
| RFU (exitation/emission at
605/670 nm) without FM64-4
|
| Empty
| 36
| 3258
|
| Blank
| 2148
| 2641
|
| 14-1
| 3571
| 4104
|
| 14-2
| 46 480
| 6181
|
| 16-1
| 3475
| 3629
|
| 16-2
| 17 430
| 4565
|
NB! Later it was discovered that the RFP that we have in the vesicles (from biobrick [http://parts.igem.org/wiki/index.php/Part:BBa_E1010 Part:BBa_E1010) has an exitation/emission peak at 584/607nm.
Colony PCR of GFP
We transferred around 15 colonies to 20µl dH2O and incubated in the PCR-machine at 95°C for 15 min. Sentrifuged for 2 min 14.000rpm. 1µl was used as template for PCR. Gel electroforeses confirm a band around 750bp which is the expected size of the GFP. DNA purification was performed using a putification kit and the concentration was measured using Nanodrop.
Gibson Assembly 3
A new gibson with the newly transformed GFP was performed. Insert and vector concentrations of 200ng, 10µl DNA 10µl Gibson Mastermix. 50°C, 15 min. A sample of 2µl was removed after 5 min. Product was incubated in -20°C immidiately to stop the reaction. 2µl transformed in ER2566 and plated. Control with all DNA was plated on Amp and Chl plates.
| Plate
| Colonies
| Concentration(ng/µl)
|
| 1
| 57
| 108.1
|
| 2
| 130
| 109.3
|
| 3
| 52
| 29.3
|
| 4
| 12
| 19.6
|
| 5
| 38
| 99.3
|
| 6
| 31
| 25.4
|
| 7
| 85
| 23.3
|
| 8
| 42
| 24
|
| Control Amp
| 95
|
| Control Chl
| 34
|
The Concentration was measured after miniprep using nanodrop. The colonies on both controlplates indicate contamination at some stage.
A new transformation was started with RFP, BB and Ptet GFP <partinfo>BBa_I13522</partinfo>, to check if the different GFP will work. Tat was isolated from ER2566 cells using colony PCR and product used as template in PCR with tat-primers. They were plated and incubated at 37°C overnight.
Wednesday 07.08.13
SDS-PAGE of vesicle samples
SDS-PAGE was run on the samples from the day before. As showed in figure 9 all of the samples had vesicles, but sample 14-2 had a higher density.
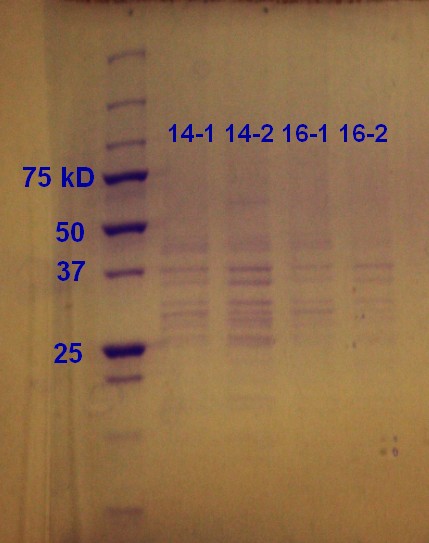 Figure 9: Ladder applied is Precision Plus ProteinTM Unstained Standards.
Figure 9: Ladder applied is Precision Plus ProteinTM Unstained Standards.
New transformation
BB, RFP, GFP and tat was transformed again to try and eliminate contamination at any possible step. The standard iGEM [http://parts.igem.org/Help:Protocols/Transformation Transformation Protocol] was used.
Another round of small-scale vesicle preparation
Step 1 and 2 of the small-scale vesicle preparation were performed with ER2566 transformed with construct from the second Gibson assembly attempt. Two colonies (see picture 8) were selected and incubated for 14 and 16 hours in step 2. The cultures were incubated aerobically in LB media with ampenicillin (100 µL/mL) in step 1 and areobically in plain LB in step 2. The samples are again labeled as 'hours incubation in step 2 - colony number'
Confirmation PCR of Gibson Construct with Taq Polymerase
We got several bands on the gel from gibson, it is unsure if they are the right size so to confirm what product was made in the gibson assembly we ran a confirmation PCR using Taq Polymerase Taq Pol Protocol.
Different combinations of primers to detect different combinations.
| Sample
| Target
| F_Primer
| R_Primer
| Tm
|
| A
| RFP
| F_l_RFP
| R_RFP
| 66
|
| B
| GFP+RFP
| F_Tat_GFP
| R_RFP
| 66
|
| C
| Tat+GFP+RFP
| F_pl.b_Tat
| R_RFP
| 63
|
| D
| GFP
| F_tat_GFP
| R_GFP
| 68
|
| E
| Tat
| F_pl.b_Tat
| R_Tat
| 63
|
| F
| Tat+GFP
| F_pl.b_Tat
| R_GFP
| 63
|
Band was visible for RFP around 750bp, so that is confirmed. Strong band for the Tat_GFP_RFP construct, but the size is only around 850bp. For it to contain tat and the 2 FPs it should be around 1600bp. It is probable that tat has attached to the RFP instead of the GFP even though the primers are not designed so that this is possible. More tests will be run.
Thursday 08.08.13
Small-scale vesicle preparation
Step 4-12 of the small-scale vesicle preparation were performed. In addition a 10-6 dilution of all the samples were made and plating of 100µL of 1 mL of the dilution on Amp-LA was performed in step 4. The plates were left in the incubator overnight. The plates with bacteria from colony 1 had growth (approximately 250-300 colonies) indicating that the bacteria still had their plasmid. The controll plate with bacteria from colony 2 had 14 colonies (14-2) and 2 colonies (16-2).
The cellmedia with bacteria from colony 1 (14-1 and 16-1) has a pink color, while the other samples did not.
Optical denisty (OD) at 600 nm was mesured in step 4 as indicated in the table below. The samples were diluted 1:10 with LB media. The true OD of 14-1 and 16-1 might be disturbed by the production of RFP, giving these samples a higher OD.
| Sample
| OD600
|
| 14-1
| 0.566
|
| 14-2
| 0.400
|
| 16-1
| 0.555
|
| 16-2
| 0.504
|
Sample 16-1 was disregarded for RFU-measurements in step 12 due to an experimental mistake.
Samples for SDS-PAGE was prepared in step 12.
RFU was measured as decribed in step 12 of the small-scale vesicle preparation protcol. As the vesicles were expected to contain RFP if the tat sequence is present, RFU was also measured without FM4-64 at exitation/emission at 605/670 (the known exitation/emission for RFP). Results were as indicated in the table below:
| Sample
| RFU (exitation/emission at
515/640 nm) with FM4-64
| RFU (exitation/emission at
584/607 nm) without FM4-64
|
| Empty
| 37
| 18 147
|
| Blank
| 1736
| 45 927
|
| 14-1
| 9030
| 40 818
|
| 14-2
| 20 745
| 31 325
|
| 16-1
|
|
|
| 16-2
| 46 126
| 37 304
|
As the highest 584/607-measurement is the blank sample, we conclude that there are no RFP present in the vesicles.
PCR
Primers for CFP (<partinfo>BBa_E0020</partinfo>) and YFP (<partinfo>BBa_E0030</partinfo>) arrived today, so PCR was run with these primers. TO avoid any contamination, we added 1 µl of Dpn1 to the amplicon and put it in a 37 °C waterbath for an hour. We used the QIAGEN PCR purification kit to isolate the DNA and measured the concentration by [http://www.nanodrop.com/library/nd-1000-v3.7-users-manual-8.5x11.pdf NanoDrop ND-1000 Spectrophotometer].
| Sample
| Concentration (ng/µl)
|
| YFP
| 64
|
| CFP
| 92,8
|
Restriction cutting of construct
2 different samples of the gibson assembly product was used in restriction analysis with 3 enzymes. Two of the enzymes cut on either side of the construct and the third cuts the GFP at 323 bp, rughly in the middle of the gene. 4 different combinations were performed. We used the restriction digest [http://parts.igem.org/Help:Protocols/Restriction_Digest protocol] from iGEM webpage.
| #
| Enzyme1
| Enzyme2
|
| 1
| EcoRI
| SpeI
|
| 2
| EcoRI
| PmlI
|
| 3
| PmlI
| SpeI
|
| 4
| BsaAI
| SpeI
|
FIND PIC OF GEL!
<center> Friday 09.08.13
SDS-PAGE of vesicle samples
SDS-PAGE was run on the samples from the day before. As showed in figure 10 the 16-2 sample seems to have the highest protein content followed by 16-1. It seems that the 16 hour incubation is to prefer when considering the SDS-PAGE (figure 9) and the RFU-measurements (table, thursday 08.08.3013)
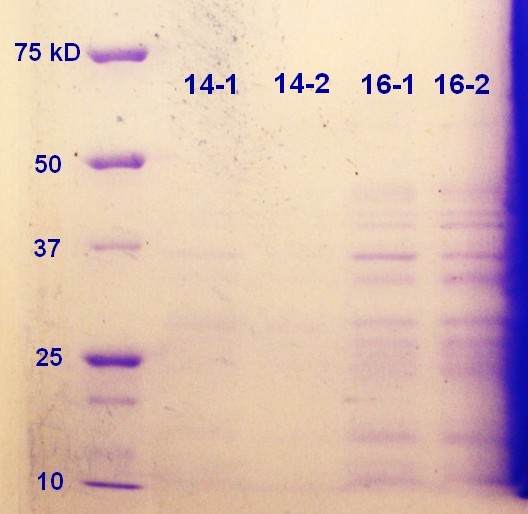 Figure 10: Ladder applied is Precision Plus ProteinTM Unstained Standards.
Figure 10: Ladder applied is Precision Plus ProteinTM Unstained Standards.
PCR
New PCR on GFP, RFP, BB and tat. Very weak band on BB. Ran new Expand high fidelity DNA polymerase kit with BB-PCRproduct as template. Still low concentration.
Gibson Assembly 4
In addition to the Tat_GFP_RFP construct(TGR) it was made a Tat_CFP_YFP construct(TCY). Tranformed(X5) and incubated overnight.
Saturday 10.08.2013
The colonies on the 10 plates were counted.
| Plate
| Colonies TCY
| Colonies TGR
|
| 1
| 59
| 45
|
| 2
| 32
| 32
|
| 3
| 62
| 39
|
| 4
| 49
| 14
|
| 5
| 100
| 20
|
The plates with the most colonies from both constructs(TCY:plate 5, TGR: plate 1) were used for colony PCR, appr. 20 colonies from each plate was transferred to 20 µl of dH2O, and incubated in the PCR for 15 min, at 95 °C. One colony from the same plates was incubated in liquid media and left overnight for miniprep the next day.
The results were disappointing again and confirmation PCR did not give the desired sizes.
Monday 12.08.2013
Gibson assembly with different construct
The experiments using tat_2xFPs_BB have been unsuccessful at this point according to our confirmation PCRs. We are therefor trying to make a 2 piece construct instead; Tat_GFPmut3* and Tat_RFP. Find GIBSON ASSEMBLY PROTOCOL!!??NBNBNBN
| Template
| Concentration(ng/µl)
| Volume in GA(µl)
|
| Tat
| 36
| 3
|
| GFPmut3*
| 58.1
| 2
|
| BB
| 39
| 5
|
Add 10 µl of gibson mastermix. Incubate for 15 min at 95 °C.
| Template
| Concentration(ng/µl)
| Volume in GA(µl)
|
| Tat
| 36
| 3
|
| RFP
| 140
| 1
|
| BB
| 39
| 5
|
Add 10 µl dH2O and 10 µl of gibson mastermix. Incubate for 15 min at 95 °C. Then both were transformed in DH5α cells.
Improving concentration of BB
The BB was run with Expand high fidelity polymerase [http://francois.schweisguth.free.fr/protocols/High_fildelity_roche.pdf Protocol] to get a higher concentration, so tht it will be better for future gibson assemblies. The annealing temperatur was set to 65 °C.
| Reactant
| µl
|
| Buffer 2
| 2
|
| dNTP
| 0.5
|
| Primers
| 0.5
|
| Template
| 1
|
| Polymerase
| 0.25
|
| dH2O
| 15.25
|
This gave an even weaker result, which is unexpected. Our next move is to use colonies of the backbone and incubate in liquid media for new miniprep and hopefully a better yield. As we are running short on purified tat that will also be incubated and run on the PCR again.
PCR with Protein G
Our Primers for protein G has arrived, as has the 5 different Streptococcus dysgalactiae ssp equisimilis samples collected from St. Olavs hospital. The strains of the bacteria samples are unknown as they are collected from patients. There are 3 different forward primers, one has the tat sequence overhang (F_tat_PrG), one has the natural sequence that the protein has in Streptococcus dysgalactiae ssp. equisimilis (F_pl.b_PrG) and one has the pelB sequence overhang (R_pl.b_pelB_PrG). There are two different reverse primers, one that will anneal to the end of the DNA sequence for the gene (R_pl.b_PrG) and another that will anneal to the end of the protein coding sequence (see [http://www.ncbi.nlm.nih.gov/nuccore/X06173 registered gene sequence]). They will be run in 6 different combinations:
| Sample
| F_Primer
| R_Primer
| Tm
|
| A
| F_pl.b_PrG
| R_pl.b_PrG
| 61
|
| B
| F_pl.b_PrG
| R_pl.b_PrGstop
| 64
|
| C
| F_tat_PrG
| R_pl.b_PrG
| 63
|
| D
| F_tat_PrG
| R_pl.b_PrGstop
| 68
|
| E
| F_pl.b_pelB_PrG
| R_pl.b_PrG
| 63
|
| F
| F_pl.b_pelB_PrG
| R_pl.b_PrGstop
|
As we have 5 different Streptococcus dysgalactiae ssp. equisimilis samples, 30 different PCR-reactions were run.
Standard conditions for Phusion PCR was used. The products was run on gel for appr. 1h. A very clear band with the size of around 1300-1400 bp was found in sample D1. This is a smaller DNA fragment compared to the [http://www.ncbi.nlm.nih.gov/nuccore/X06173 known gene sequence] of about 1700 bp. Since the size of Protein G is known to vary among the different strains, we can still conclude that our PCR-product is Protein G. Bacterial sample 1 using the D-primers(F_tat_PrG + R_pl.b_PrGstop) is most likely Protein G with the tat-overhang. The next step is to use this product in gibson assembly with tat and BB.
Tuesday 13.08.2013
Restriction digest of BB
To remove any contaminants in our BB sample we are running a restriction digest with only one enzyme, HindIII, and cutting the correct band from the gel. The band was purified with the (FIND PROTOCOL).
PEC
To link two PCR products together we used a tecnique called PEC-Polymerase extension cloning. This method uses the established overhangs of the fragments to link them toghether in the PCR using only the polymerase(phusion). And then after some 10 cycles the program is paused and primers for the whole construct is added to amplify that.
The first experiment using SLIC: To link toghether tat_GFP and tat_GFPmut3*. A two-step PCR program was setup with 10 cycles in the first step and 20 cycles in the second step. For the first step the flanking(overlapping) sequences were used to determine annealing temperature = 72 %deg;C, and for the second step only the primers without the flanking sequence were used to calculate anealingtemperature = 62 %deg;C. Other than that standard PCR conditions using phusion polymerase was used. (RESULTS FROM THIS????)
CPEC
CPEC-circular polymerase extension cloning is the same principle as PEC, only circular. Du to this there is no need for primers, only that the overhans all fit to each other. We attempted this method with our to FP constructs. Tat_GFP_RFP and Tat_CFP_YFP- (ELLEN: Could you add how you calculated the amount of each fragment)
| Reactant
| Volume(µl)
| Concentration(ng/µl)
|
| HF-buffer
| 4
|
|
| dNTP
| 1.6
|
|
| Phusion polymerase
| 0.2
|
|
| Vector BB
| 4
| 50
|
| GFP
| 1
| 88.4
|
| RFP
| 0.5
| 140
|
| Tat
| 1.7
| 36.5
|
| CFP
| 1
| 92.8
|
| YFP
| 1
| 64
|
| dH2O
| up tp 20
|
One reaction with Tat_GFP_RFP_BB and one with Tat_CFP_YFP_BB. (I dont have the PCR conditions used here!!ANYONE??).
Wednesday 14.08.2013
PCR with TCY construct
Standars phusion polymerase protocol with a 20 µl reaction. (FIND PIC)
PCR to link tat and GFP
As the PEC reaction did not confirm a link, were trying the same using PCR. Phusion polymerase 20 µl reaction. Annealing temperature 72/62 in 2-step PCR.(FIND PIC).
Restriction digest of tGR and tCY product from CPEC(13.08.13)
(The standard iGEM [http://parts.igem.org/Help:Protocols/Restriction_Digest Restriction Digest Protocol] was used with SpeI enzyme. The sample was then run on a gel(FIND PIC). Strong band at appr. 2300 bp indicates the right size, but also some contamination is visible in the samples.) INGRID!, I think what happen here is what I describe under
The standard iGEM [http://parts.igem.org/Help:Protocols/Restriction_Digest Restriction Digest Protocol] was used applied with SpeI enzyme on the tGR and tCY products from yesterday (13.08.13). The digested samples were then run on a gel together with a previous PCR-product of BB (see figure below) that served as a referance.
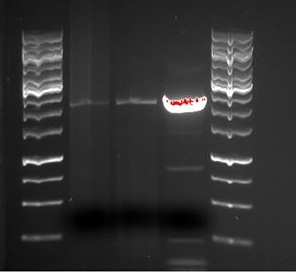 figure: From the left; tGR, tCY and BB (PCR product).
figure: From the left; tGR, tCY and BB (PCR product).
As indicated in the picture, there does not seem to be any tGR or tCY construct as the restricted products are the same size as the BB when they should be longer. There also seems to be a bit of contamination in the BB sample.
Thursday 15.08.2013
Purify BB by running it on gel
To remove any contaminants from the BB sample we ran 50 µl og BB sample on a gel, cut it and purified it using neb (FIND PROTOCOL).
CPEC with purified BB and tat_PrG
The same conditions as last time were used. BB: 4 µl and tat: 3µl. Phusion 20 µl reaction.
| Step
| Temperature(°C)
| Time(sec)
|
| Initial denaturation
| 98
| 30
|
| Denaturation
| 98
| 15
|
| Annealing
| 52
| 30
|
| Elongation
| 72
| 110
|
| Final Elongation
| 72
| 5 min
|
The product was transformed into DH5α cells and ER2566 cells.
Direct transformation(DT) of BB and Tat_PrG
1:1 Ratio, BB: 2 µl, Tat_PrG: 1.5 µl. Directly transferred into DH5α cell and ER2566 cells
SLIC with BB and Tat_PrG
| Reactant
| Volume
|
| NEB Buffer
| 1
|
| BSA
| 1
|
| T4 polymerase
| 0.2
|
| BB vector
|
|
| Tat_PrG
|
|
| dH2O
|
|
Incubated in room tempreature for 2.5 minutes, then 10 min on ice and transformed into DH5α cell and ER2566 cells.
These are the transformations done today:
| Sample
| Method
| Cells
|
| 1
| CPEC
| DH5α
|
| 2
| CPEC
| ER2566
|
| 3
| DT
| DH5α
|
| 4
| DT
| ER2566
|
| 5
| SLIC
| DH5α
|
| 6
| SLIC
| ER2566
|
| 7
| Control BB
| DH5α
|
| 8
| Control Tat_PrG
| DH5α
|
Thursday 15.08.2013
Purify BB by running it on gel
To remove any contaminants from the BB sample we ran 50 µl og BB sample on a gel, cut it and purified it using neb (FIND PROTOCOL).
CPEC with purified BB and tat_PrG
The same conditions as last time were used. BB: 4 µl and tat: 3µl. Phusion 20 µl reaction.
| Step
| Temperature(°C)
| Time(sek)
|
| Initial denaturation
| 98
| 30
|
| Denaturation
| 98
| 15
|
| Annealing
| 52
| 30
|
| Elongation
| 72
| 110
|
| Final Elongation
| 72
| 5 min
|
The product was transformed into DH5α cells and ER2566 cells.
Direct transformation(DT) of BB and Tat_PrG
1:1 Ratio, BB: 2 µl, Tat_PrG: 1.5 µl. Directly transferred into DH5α cell and ER2566 cells
SLIC with BB and Tat_PrG
| Reactant
| Volume
|
| NEB Buffer
| 1
|
| BSA
| 1
|
| T4 polymerase
| 0.2
|
| BB vector
|
|
| Tat_PrG
|
|
| dH2O
|
|
Incubated in room tempreature for 2.5 minutes, then 10 min on ice and transformed into DH5α cell and ER2566 cells.
These are the transformations done today:
| Sample
| Method
| Cells
|
| 1
| CPEC
| DH5α
|
| 2
| CPEC
| ER2566
|
| 3
| DT
| DH5α
|
| 4
| DT
| ER2566
|
| 5
| SLIC
| DH5α
|
| 6
| SLIC
| ER2566
|
| 7
| Control BB
| DH5α
|
| 8
| Control Tat_PrG
| DH5α
|
New primers and PCR
New primers arrived for BB, tat and YFP. The new primers (except YFP primer) has overhengs where the equivalent primers had not. A new reverse primer for YFP was designed because the old one lacked the terminator (TAATAA) sequence New PCR was run with standard phusion protocol 50 µl reaction.(FIND PIC)
| Primer name
| Target
|
| R_GFP_tat
| tat
|
| R_CFP_tat
| tat
|
| F_GFP_pl.b
| BB or pl.b
|
| R_CFP_pl.b
| BB or pl.b
|
| R_pl.b_YFPnew
| YFP
|
Friday 16.08.2013
Redoing PCR form yesterday
The PCR was negative for sample 1 and 4 yesterday, they were ran again and gave bands. (FIND PIC)
Gibson with new TGR overhangs
| Reactant
| volume
|
| BB
| 2.5
|
| Tat
| 0.3
|
| GFP
| 0.7
|
| RFP
| 0.45
|
| Gibson mastermix
| 15
|
The products were transformed and plated. A control for tat and BB was also plated.
SLIC with new overhangs
| Reactant
| Concentration
| volume
|
| BB vector
|
|
|
| Tat
|
|
|
| GFP
|
|
|
| RFP
|
|
|
Transformed and plated.
PEC with new overhangs
| Reactant
| volume
| Concentration
|
| HF-buffer
| 4
|
|
| dNTP
| 1.6
|
|
| Phusion Pol
| 0.2
|
|
| Tat
| 1
|
|
| GFP
| 1.5
|
|
| RFP
| 1
|
|
| dH2O
| 9.3
|
|
(this one gave no construct)
Confirmation PCR on gibson
It was done a confirmation PCR on the remaining sample from gibson(before transformation) using F_pl.b_tat and R-pl.b_RFP with Tm=72 °. (FIND PIC)
Saturday 17.08.2013
Minipreping of Protein G samples
A total of 18 liquid media samples containing tat_ProteinG plasmids were minipreped. In the table below there is an overview of the samples:
Table: C = CPEC, S = SLIC and DT = direct transformation.
| Sample
| Marking
| Sample
| Marking
|
| ProteinG 1
| C (1)
| Protein 10
| DT (3)
|
| ProteinG 2
| S (1)
| Protein 11
| C (4)
|
| ProteinG 3
| DT (1)
| ProteinG 12
| DT (4)
|
| ProteinG 4
| C (2)
| ProteinG 13
| C (5)
|
| ProteinG 5
| S (2)
| ProteinG 14
| DT (6)
|
| ProteinG 6
| DT (2)
| ProteinG 15
| DT (7)
|
| ProteinG 7
| C (3)
| ProteinG 16
| DT (8)
|
| ProteinG 8
| S (3)
| ProteinG 17
| DT (9)
|
| ProteinG 9
| DT (3)
| ProteinG 18
| DT (10)
|
Confirmation PCR
INGRID&ELLEN: I think this is where you guys did a mistake when you though you were doing conf. PCR on tCY/tGR but infact it was ProteinG. The correct proteinG pcr was run on sunday
On Gibson TGR 1+2 and TCY, tat_PrG CPEC Samples 1-5, tat_PrG SLIC Samples 1-3, tat_PrG DT Samples 1-10. Ran on gel (FIND PIC)
Colonies from plates with TGR, TCY Gibson and SLIC were transferred to liquid media.
EVERYTHING THAT WAS DONE DURING THIS WEEKEND IS IN SOMEONE ELSES LABJOURNAL :) FILL IN PLEASE.. Im pretty sure we did some things!
Sunday 18.08.2013
Miniprep
The Gibson and SLIC with tGR and tCY that were transerred to liquid media yesterday were minipreped.
Conformation PCR on tat_ProteinG
We did a confirmation PCR of all the 18 Protein_G plasmids that were minipreped the day before (17.08.2013). Them we ran the PCR product on a gel (see figure below)
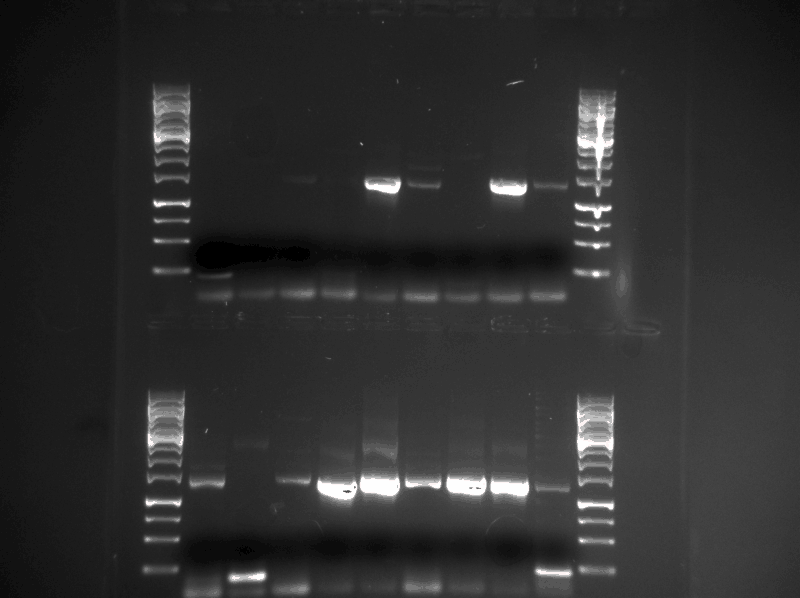 figure: First row contains ProteinG samples 1-9, second row contains ProteinG samples 10-18 (see table from saturday 17.08.2013).
figure: First row contains ProteinG samples 1-9, second row contains ProteinG samples 10-18 (see table from saturday 17.08.2013).
Thursday 22.08.2013
Samples (see table below) were prepared and sent for sequencing. The DNA fragments (tat_GFP and tat_GFPmut3) were sent with their respectful reverse primer and the plasmids (the rest of the samples) were sent with the standard
iGEM forward sequencing primer. The samples were made with the concentration between 80-100 ng/µl. The samples with to low concentration was evaporated to get higher concentration.
| Sample#
| Sample name
|
| 1
| tat_GFP
|
| 2
| tat_GFPmut3
|
| 3
| SYFP
|
| 4
| BFP
|
| 5
| ProteinG (DT8)
|
| 6
| ProteinG (S3)
|
| 7
| tGR 1 (minipreped 18.08.13)
|
| 8
| tGR 2 (minipreped 18.08.13)
|
| 9
| ER1 (see figure 8)
|
| 10
| ER2 (see figure 8)
|
Saturday 24.08.2013
PEC
We did a PEC reaction with:
1. Tat_GFP + RFP, Tm= 72/72 °C (F_pl.b_tat + R_pl.b_RFP)
2. Tat + CFP, Tm= 58/72 °C (F_pl._tat + R_l_CFP)
Same procedure as last time, 20 µl reaction, 1 µl primers added after 10 cycles. (FIND PIC)
Confirmation PCR
| Sample
| Target
|
| 1
| Gibson TGR 1
|
| 2
| Gibson TGR 2
|
| 3
| SLIC TGR 1
|
| 4
| SLIC TGR 2
|
| 5
| PEC TGR
|
| 6
| PEC Tat_CFP
|
| 7
| Control CFP
|
(FIND PIC)
Tuesday 27.08.2013
Today we got the sequencing results on the plasmids/DNA fragments we sent last week. The figures below show the alignments of the samples (always first line) and the expected sequences (always second line). Red indicates proper alignment, blue indicates mismatches
 figure: Aligment of the tat_GFP DNA fragment. The GFP part of the sequence is correct, but we do not know is the tat sequence is correct as this was not included in the sequencing results.
figure: Aligment of the tat_GFP DNA fragment. The GFP part of the sequence is correct, but we do not know is the tat sequence is correct as this was not included in the sequencing results.
 figure: Aligment of the tat_GFPmut3 DNA fragment. The GFPmut3 part of the sequence is correct, but we do not know is the tat sequence is correct as this was not included in the sequencing results.
figure: Aligment of the tat_GFPmut3 DNA fragment. The GFPmut3 part of the sequence is correct, but we do not know is the tat sequence is correct as this was not included in the sequencing results.
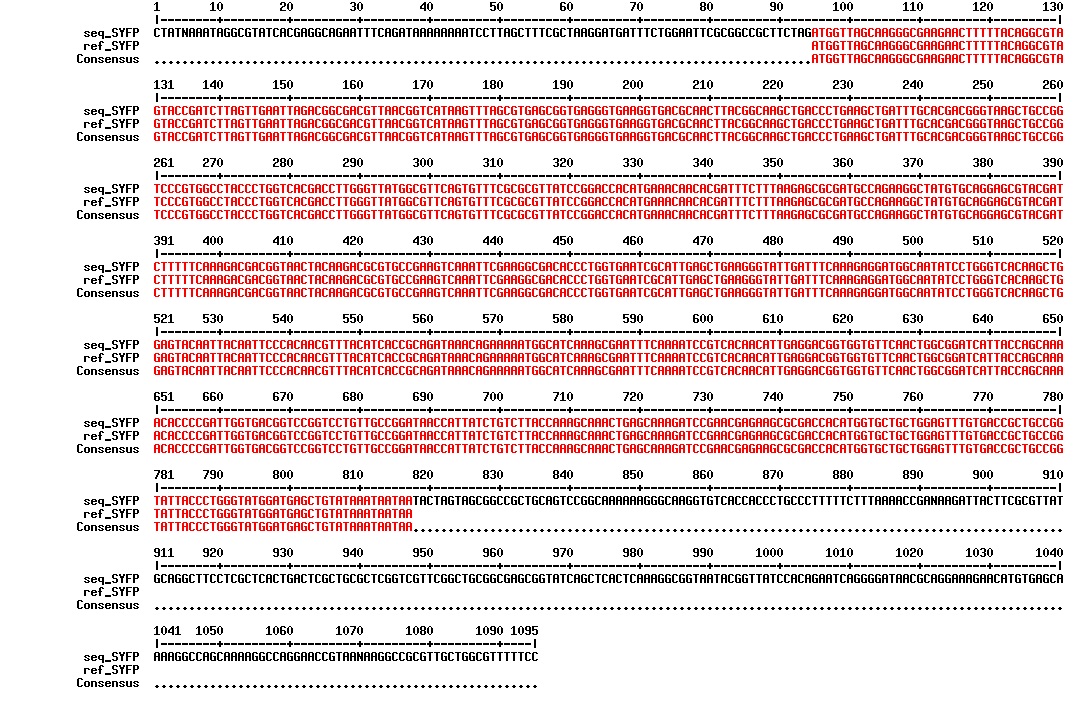 figure: Aligment of SYFP. The SYFP biobrick has the correct sequence and it is highly unknown why our primers did not work in PCR amplification of the gene.
figure: Aligment of SYFP. The SYFP biobrick has the correct sequence and it is highly unknown why our primers did not work in PCR amplification of the gene.
 figure: Aligment of SYFP. The BFP biobrick has the correct sequence and it is highly unknown why our primers did not work in PCR amplification of the gene.
figure: Aligment of SYFP. The BFP biobrick has the correct sequence and it is highly unknown why our primers did not work in PCR amplification of the gene.
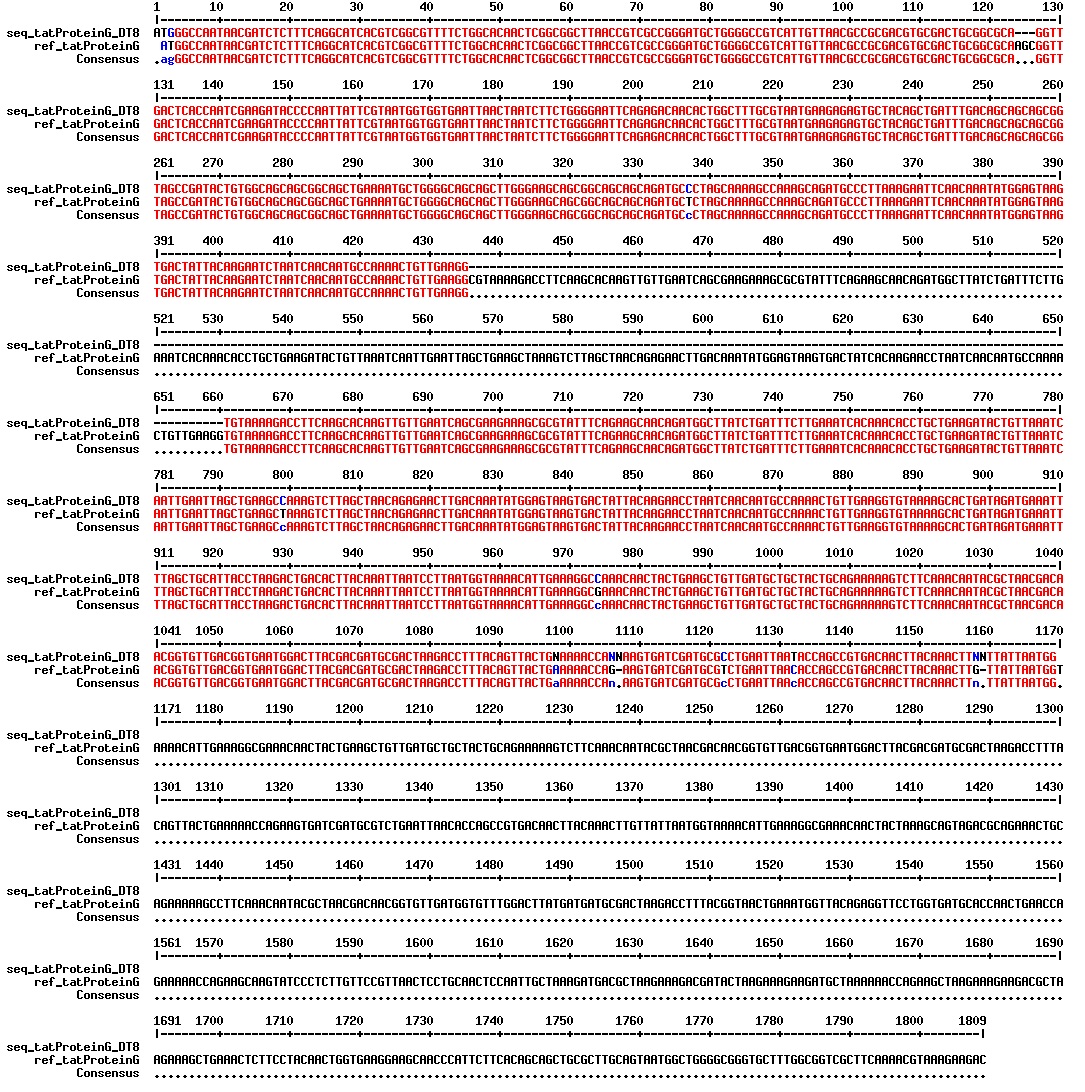 figure: Aligment of tat_ProteinG DT8. There is one segment missing in our ProteinG, as is expected as the ProteinG variant that we are dealing with is a shorter than the [http://www.ncbi.nlm.nih.gov/nuccore/X06173 registered gene sequence]. There is how ever an addition of a guanin residue in the beginning of the tat-sequence, which will cause a frameshift resulting in a non-functional protein product.
figure: Aligment of tat_ProteinG DT8. There is one segment missing in our ProteinG, as is expected as the ProteinG variant that we are dealing with is a shorter than the [http://www.ncbi.nlm.nih.gov/nuccore/X06173 registered gene sequence]. There is how ever an addition of a guanin residue in the beginning of the tat-sequence, which will cause a frameshift resulting in a non-functional protein product.
 figure: Aligment of tat_ProteinG DT8. There is one segment missing in our ProteinG, as is expected as the ProteinG variant that we are dealing with is a shorter than the [http://www.ncbi.nlm.nih.gov/nuccore/X06173 registered gene sequence]. There is how ever an deletion of a guanin residue in the beginning of the tat-sequence, which will cause a frameshift resulting in a non-functional protein product.
figure: Aligment of tat_ProteinG DT8. There is one segment missing in our ProteinG, as is expected as the ProteinG variant that we are dealing with is a shorter than the [http://www.ncbi.nlm.nih.gov/nuccore/X06173 registered gene sequence]. There is how ever an deletion of a guanin residue in the beginning of the tat-sequence, which will cause a frameshift resulting in a non-functional protein product.
 figure: Aligment of tat_GFP_l_RFP (tGR 1). The aligment is just nonsense. This plasmid does not have the intended sequence.
figure: Aligment of tat_GFP_l_RFP (tGR 1). The aligment is just nonsense. This plasmid does not have the intended sequence.
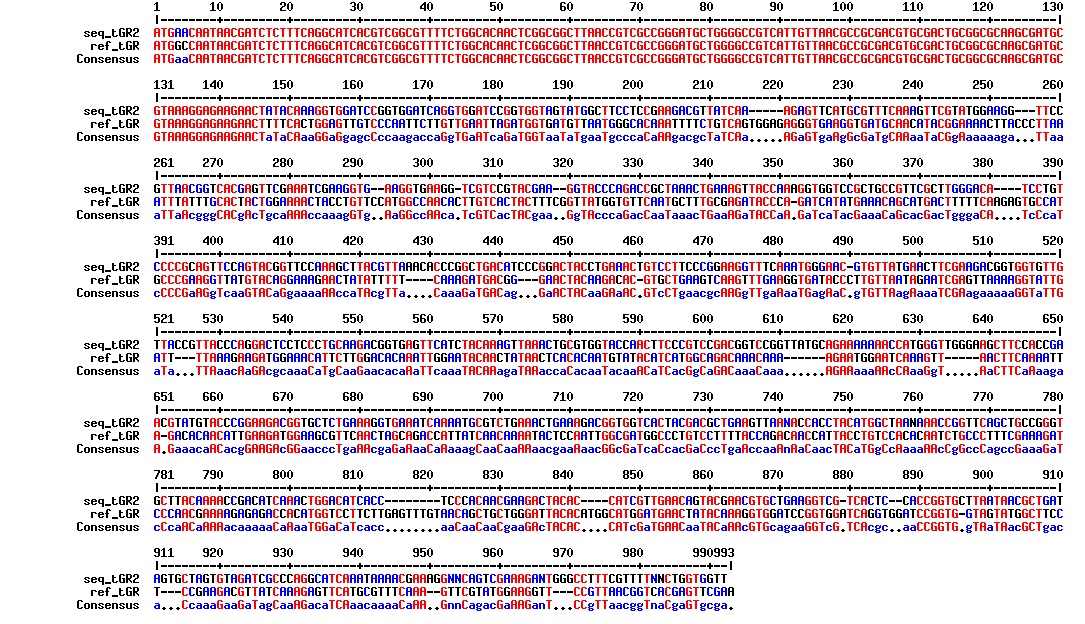 figure: Aligment of tat_GFP_l_RFP (tGR 2). The aligment is just nonsense. This plasmid does not have the intended sequence.
figure: Aligment of tat_GFP_l_RFP (tGR 2). The aligment is just nonsense. This plasmid does not have the intended sequence.
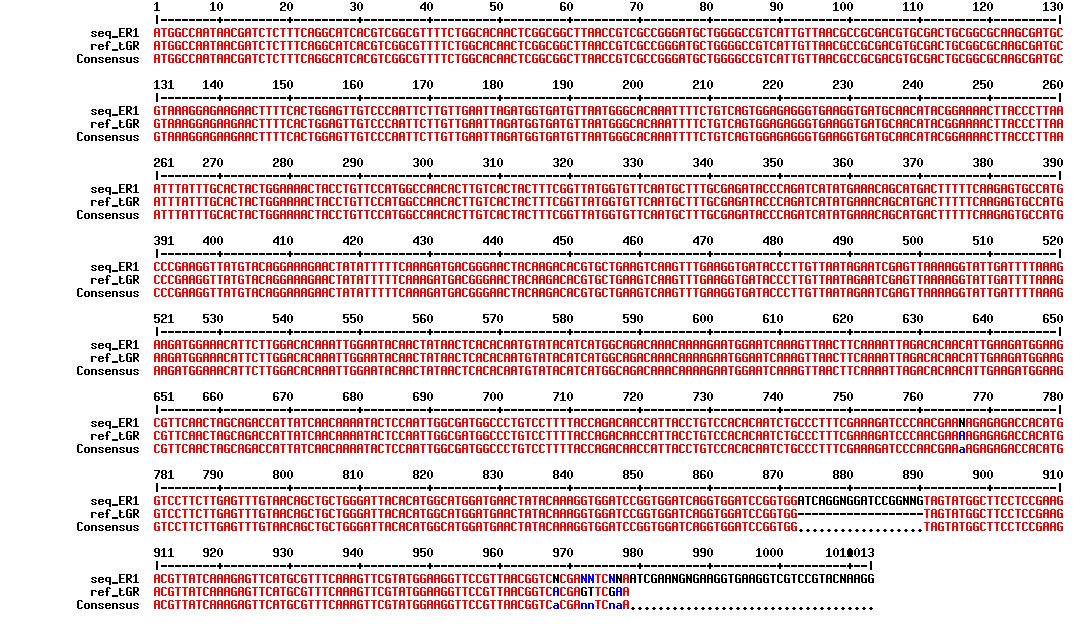 figure: Aligment of tat_GFP_l_RFP (ER1). The sequence align almost perfectly. There seems to be some sort of extra insert at the linker region, but this insert is dividable by 3, so the reading frame is maintained. This is supported by the fact that the colonies with this construct is red (see figure 8).
figure: Aligment of tat_GFP_l_RFP (ER1). The sequence align almost perfectly. There seems to be some sort of extra insert at the linker region, but this insert is dividable by 3, so the reading frame is maintained. This is supported by the fact that the colonies with this construct is red (see figure 8).
 figure: Aligment of tat_GFP_l_RFP (ER2). The aligment is just nonsense. This plasmid does not have the intended sequence.
figure: Aligment of tat_GFP_l_RFP (ER2). The aligment is just nonsense. This plasmid does not have the intended sequence.
Thursday 27.08.2013
Preparing Protein G and tCY construct for sequencing
All the rest of the samples with tat_protein G that had a positive result on the PCR confrmation (see figure on sunday 18.08.2013) was prepared and sent for sequencing. The samples were made with the concentration between 80-100 ng/µl. The samples with to low concentration was evaporated to get higher concentration.
| Sample
| Name
| Seq.ID
|
| 1
| ProteinG (S2)
| J83
|
| 2
| ProteinG (C4)
| J84
|
| 3
| ProteinG (C5)
| J85
|
| 4
| ProteinG (DT2)
| J86
|
| 5
| ProteinG (DT3)
| J87
|
| 6
| ProteinG (DT5)
| J89
|
| 7
| ProteinG (DT6)
| J88
|
| 8
| ProteinG (DT7)
| J90
|
| 9
| ProteinG (DT9)
| J91
|
| 10
| ProteinG (DT10)
| J92
|
| 11
| GIBSON TCY1
| J93
|
| 12
| GIBSON TCY2
| J94
|
| 13
| GIBSON TCY3
| J95
|
| 14
| SLIC TCY
| J96
|
Thursday 29.08.2013
Confirmation PCR on sequenced construct
The sequencing confirmed our construct: Tat_GFP_RFP. It was confirmed in one of our previous Gobson assemblies by sequencing. We therefore wanted to run a PCR to confirm the results since we know that the construct is there. The PCR was run with F_pl.b_tat and R_pl.b_RFP and the PCR products were run on a gel (see figure below):
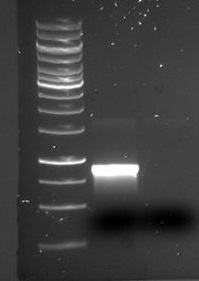 figure: Gel of conformation PCR on ER1 (tGR) construct.
figure: Gel of conformation PCR on ER1 (tGR) construct.
There is a distinct band below 1000 bp that we are not certain what is. There is also a visible band at around 1700 bp, which is the expected size of the tGR construct.
Freezing of tat_GFP_RFP construct
Bacteria with the tat_GFP_RFP construct that had a positive sequencing result (the ER1 plasmid) was prepared for freezing.
5 cryotubes: 800 µl of cell media(overnight incubated colonies of construct)
400 µl of 60% glycerol
Stored in -80 freezer(Rack 6, Shelf 7: pink box marked iGEM).
Tuesday 03.09.2013
Today we got back the sequencing results from last week (see Thursday 29.08.2013). The figures below show the alignments of the samples (always first line) and the expected sequences (always second line). Red indicates proper alignment, blue indicates mismatches.
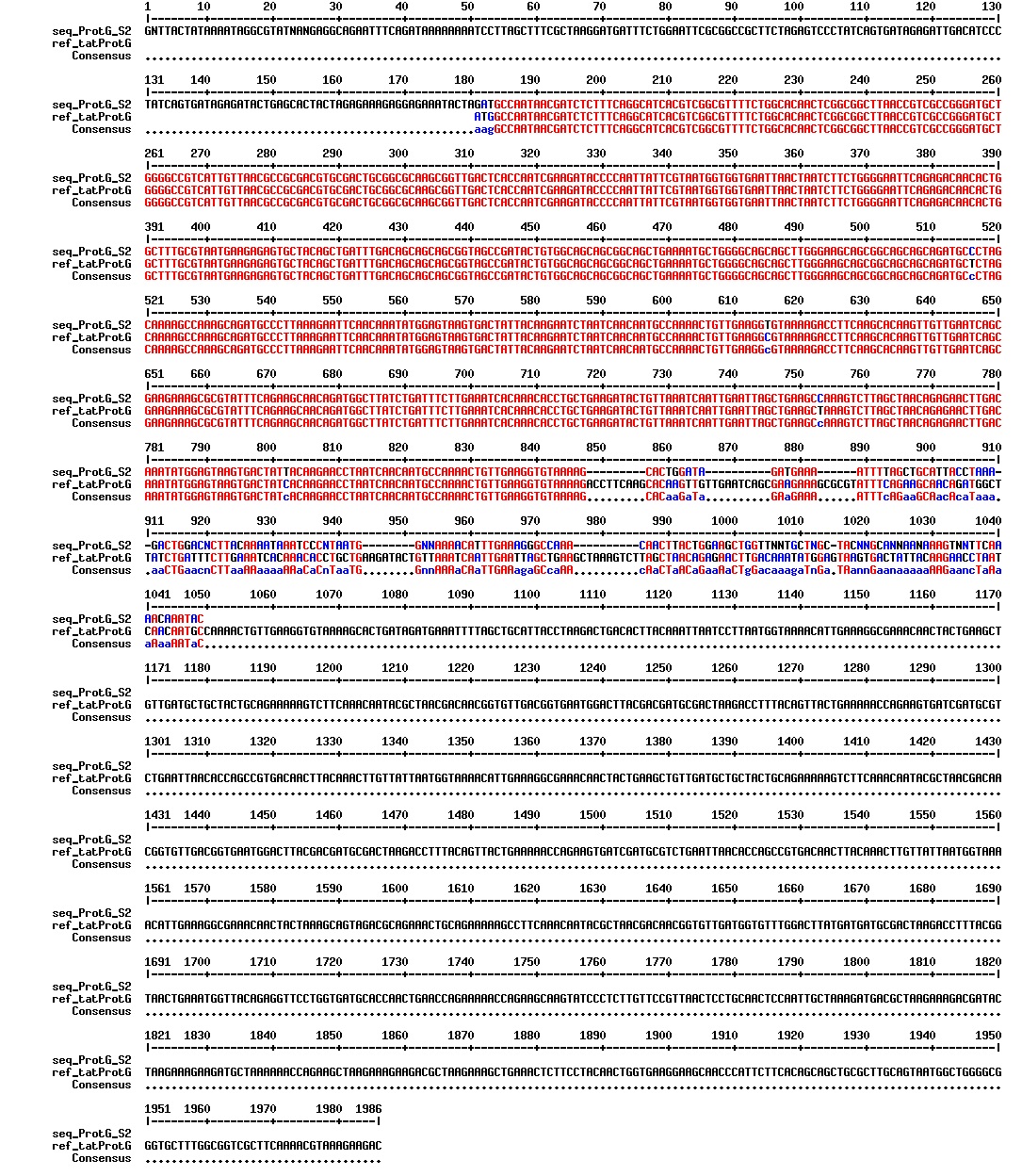 figure: Alignment of tat_ProteinG S2. As the last time there seems to be a deletion on a guanine residue that shifting the reading frame. In addition the last part of the sequence does not seem to match like S3 and D8 did.
figure: Alignment of tat_ProteinG S2. As the last time there seems to be a deletion on a guanine residue that shifting the reading frame. In addition the last part of the sequence does not seem to match like S3 and D8 did.
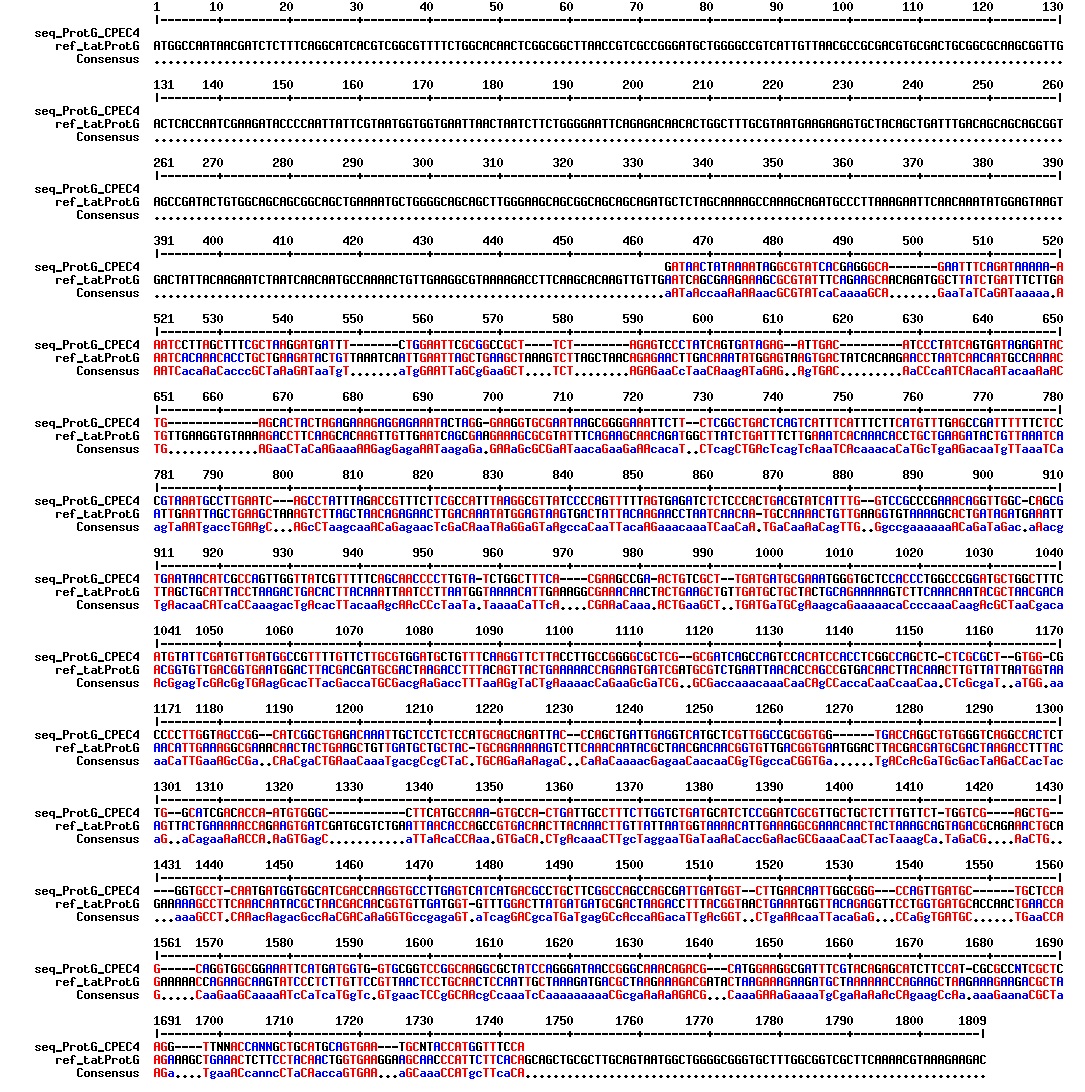
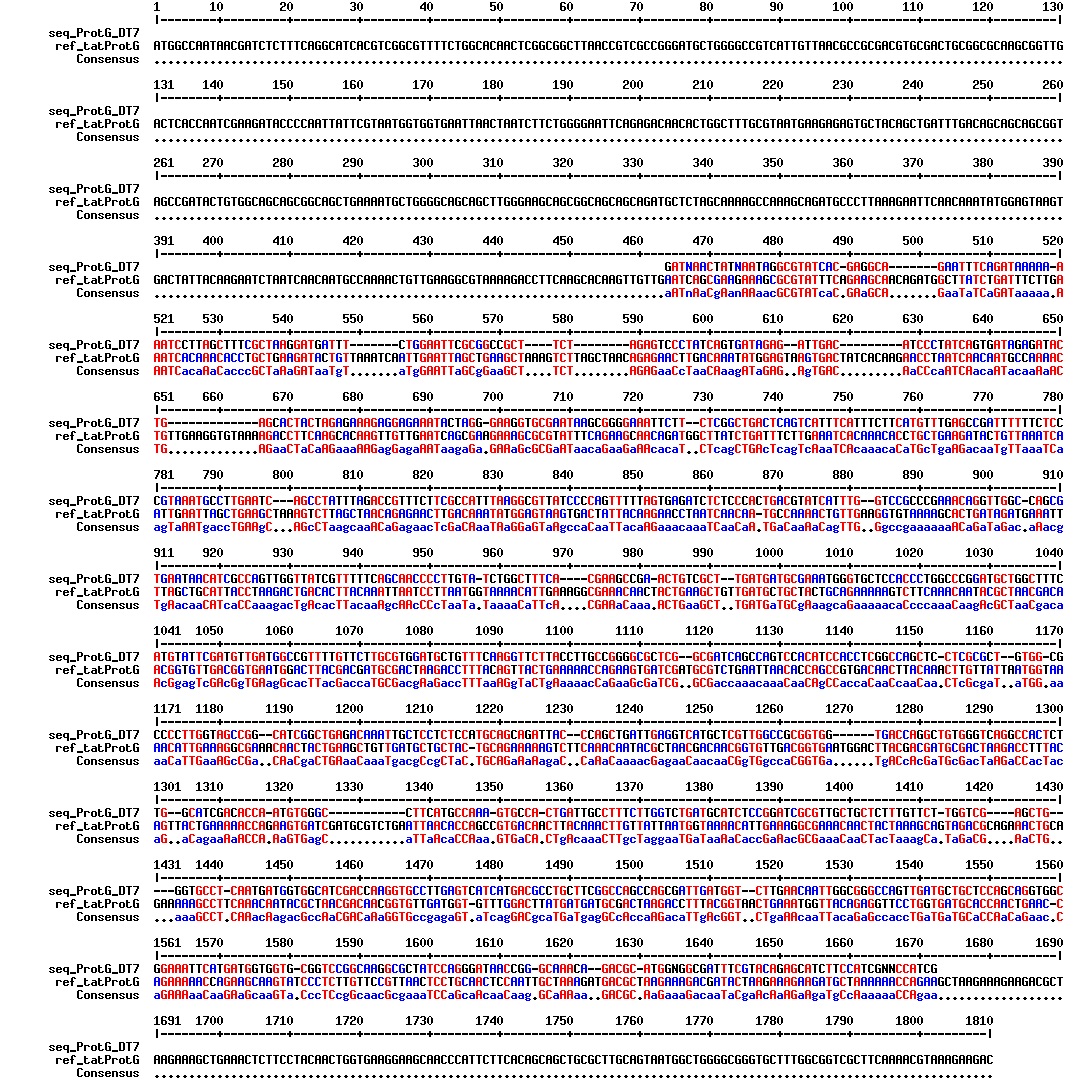
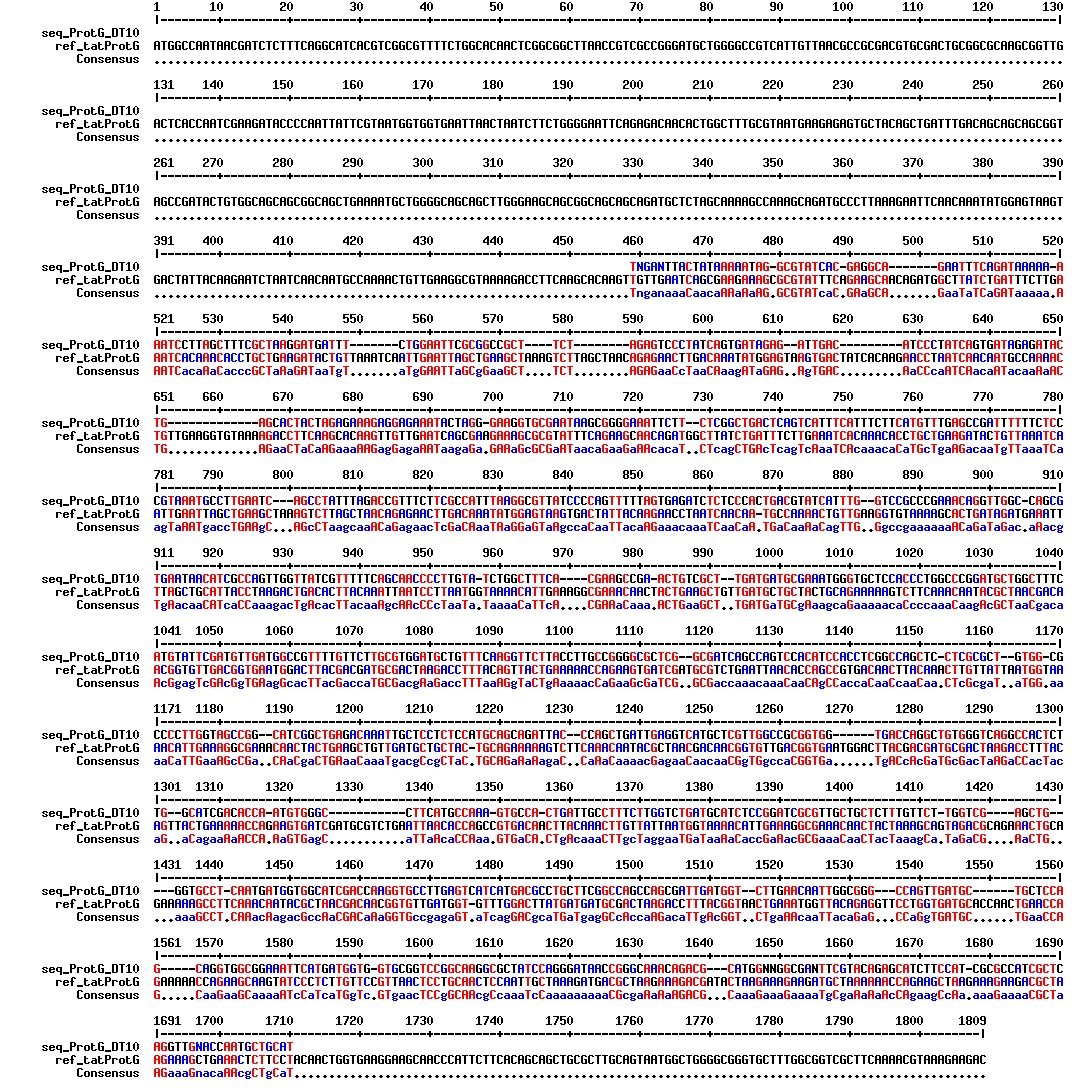 figures: Alignment of tat_ProteinG C4, tat_ProteinG C5, tat_ProteinG DT2, tat_ProteinG DT7 and tat_ProteinG DT10. None of these sequences makes any sense. It is safe to conclude that ProteinG is not in these sequences.
figures: Alignment of tat_ProteinG C4, tat_ProteinG C5, tat_ProteinG DT2, tat_ProteinG DT7 and tat_ProteinG DT10. None of these sequences makes any sense. It is safe to conclude that ProteinG is not in these sequences.
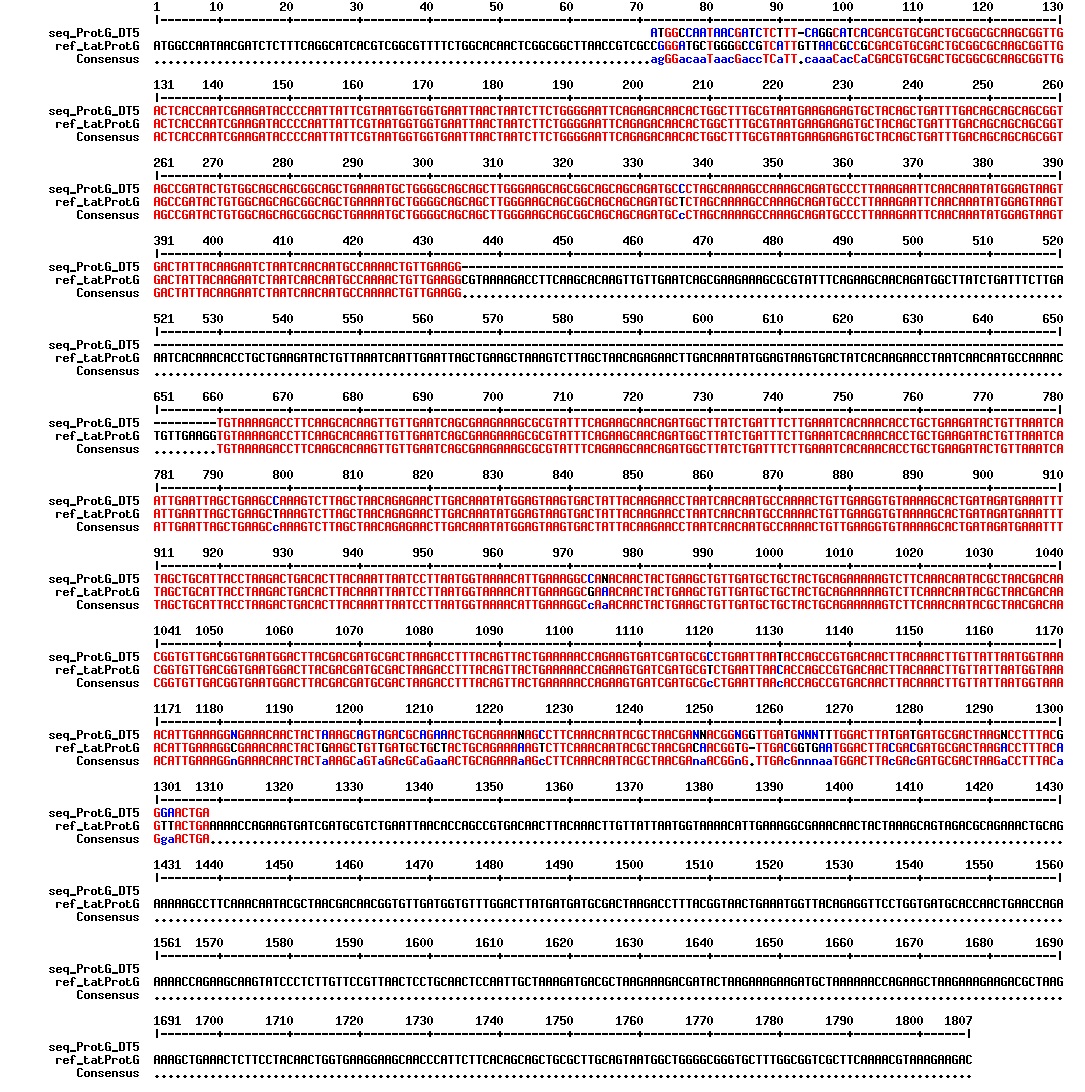
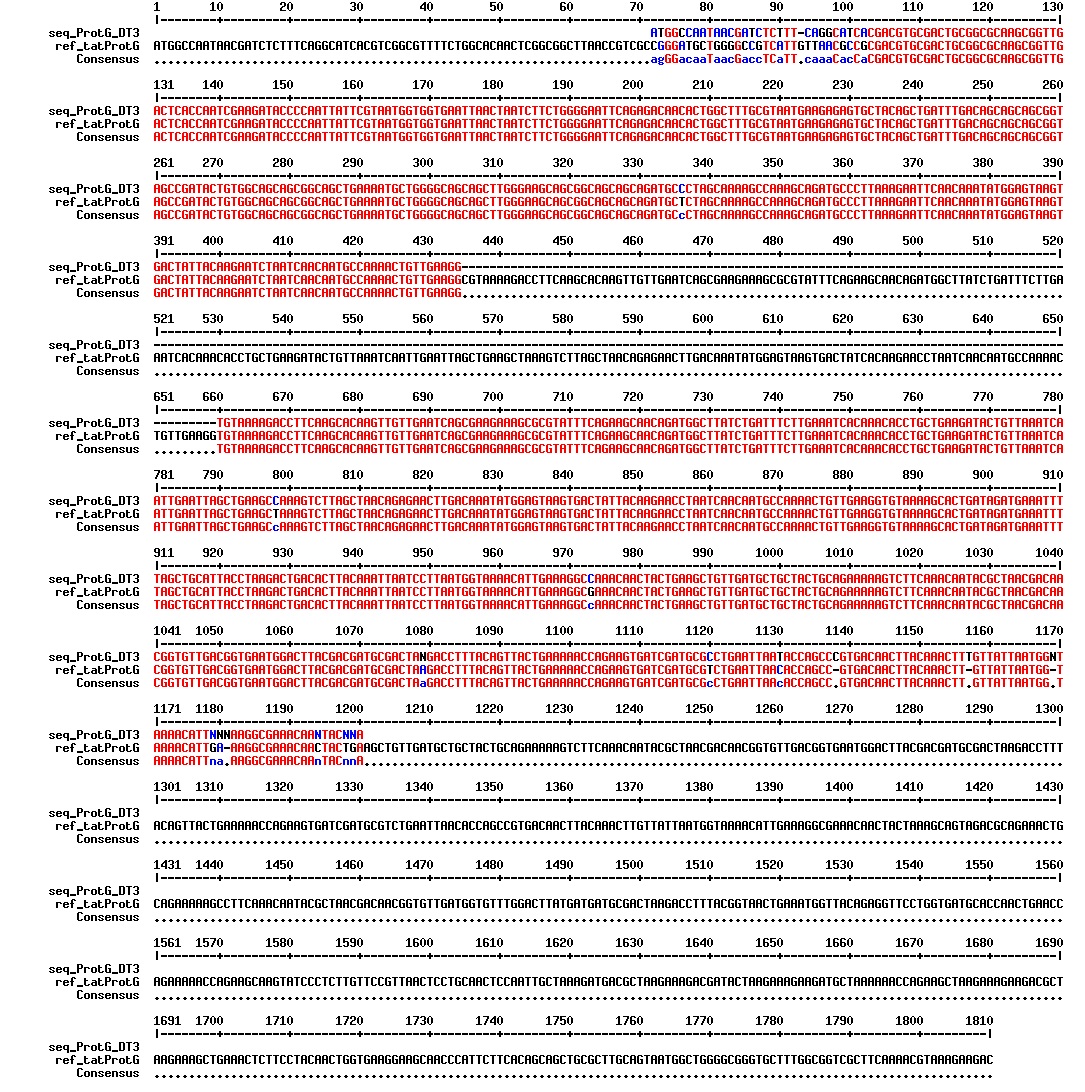
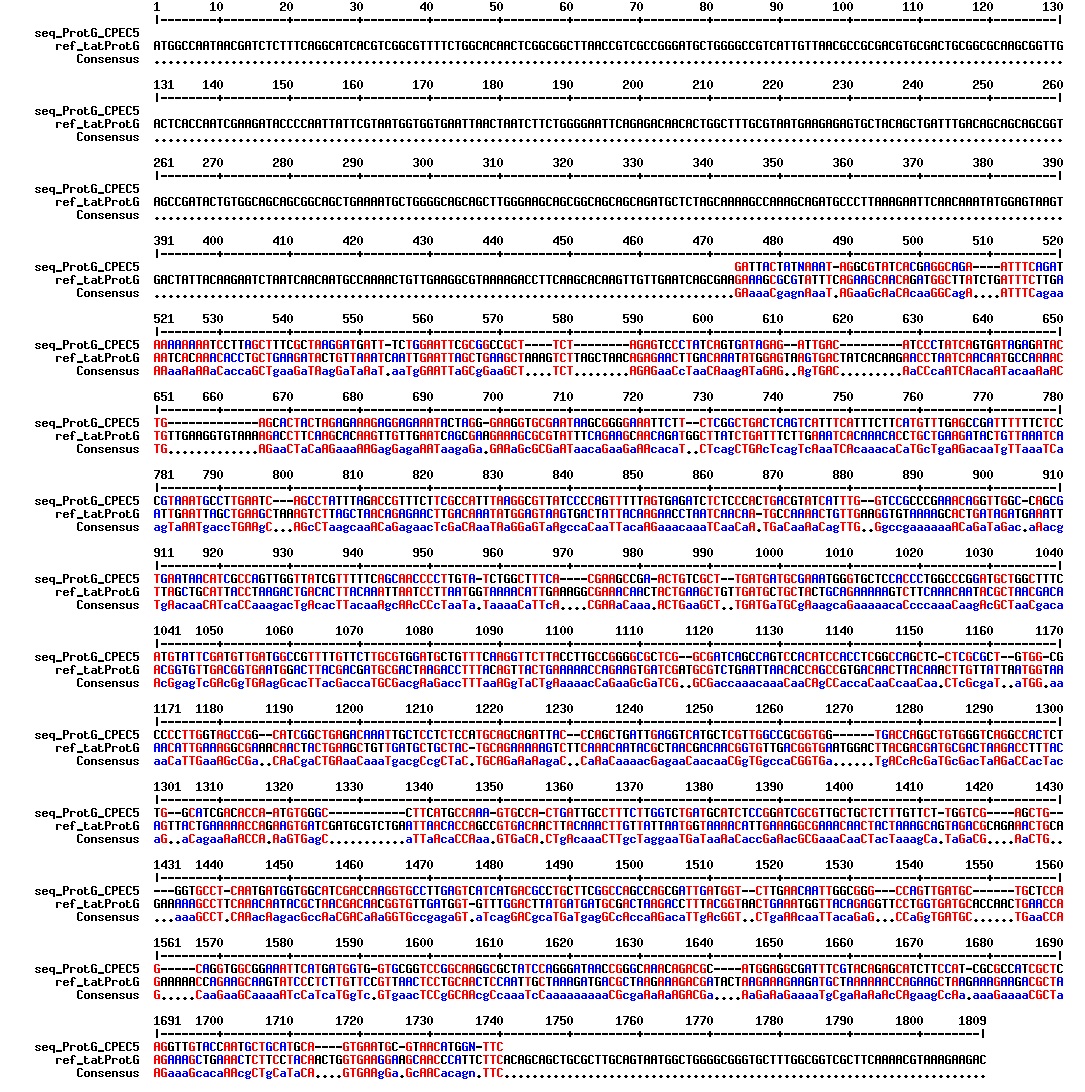
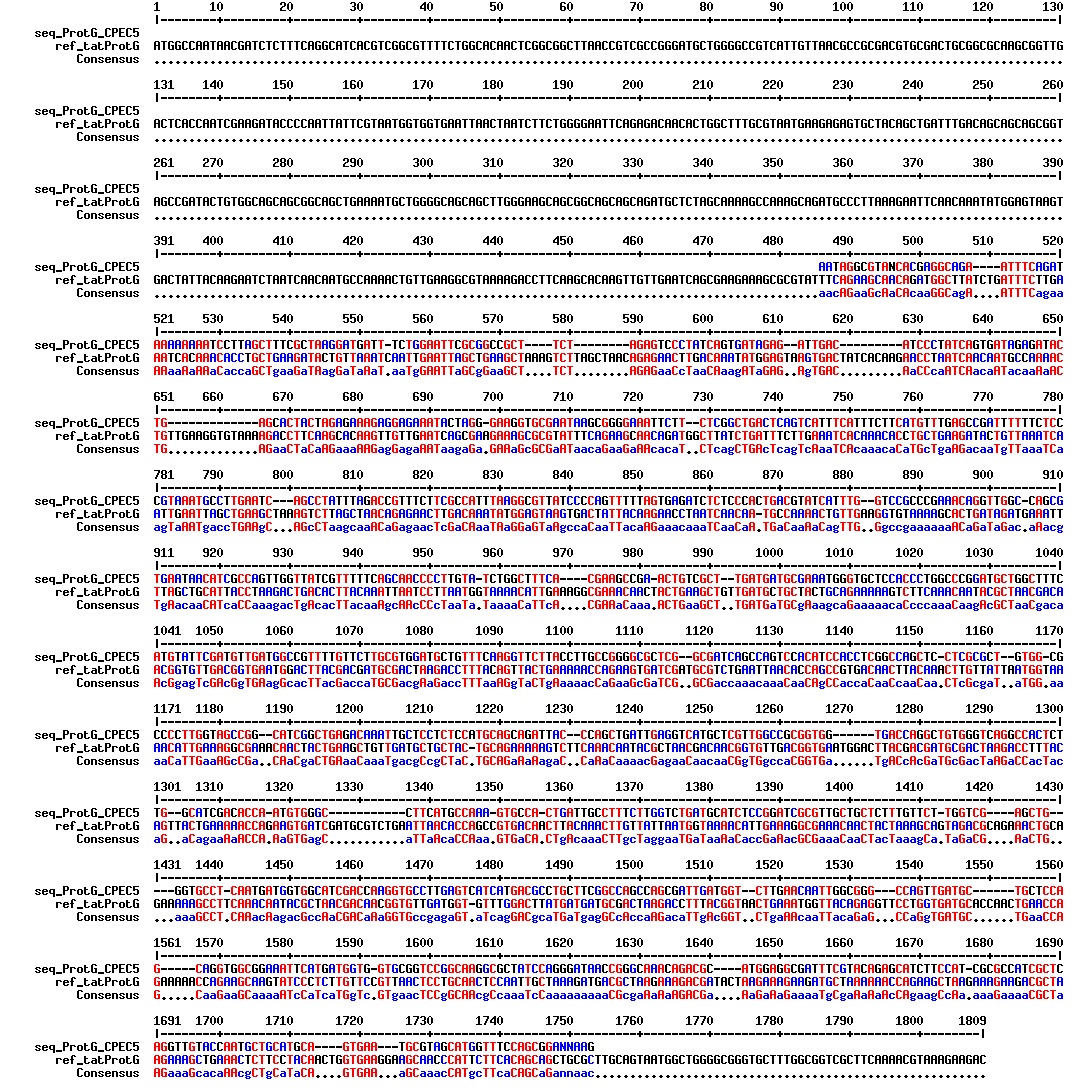
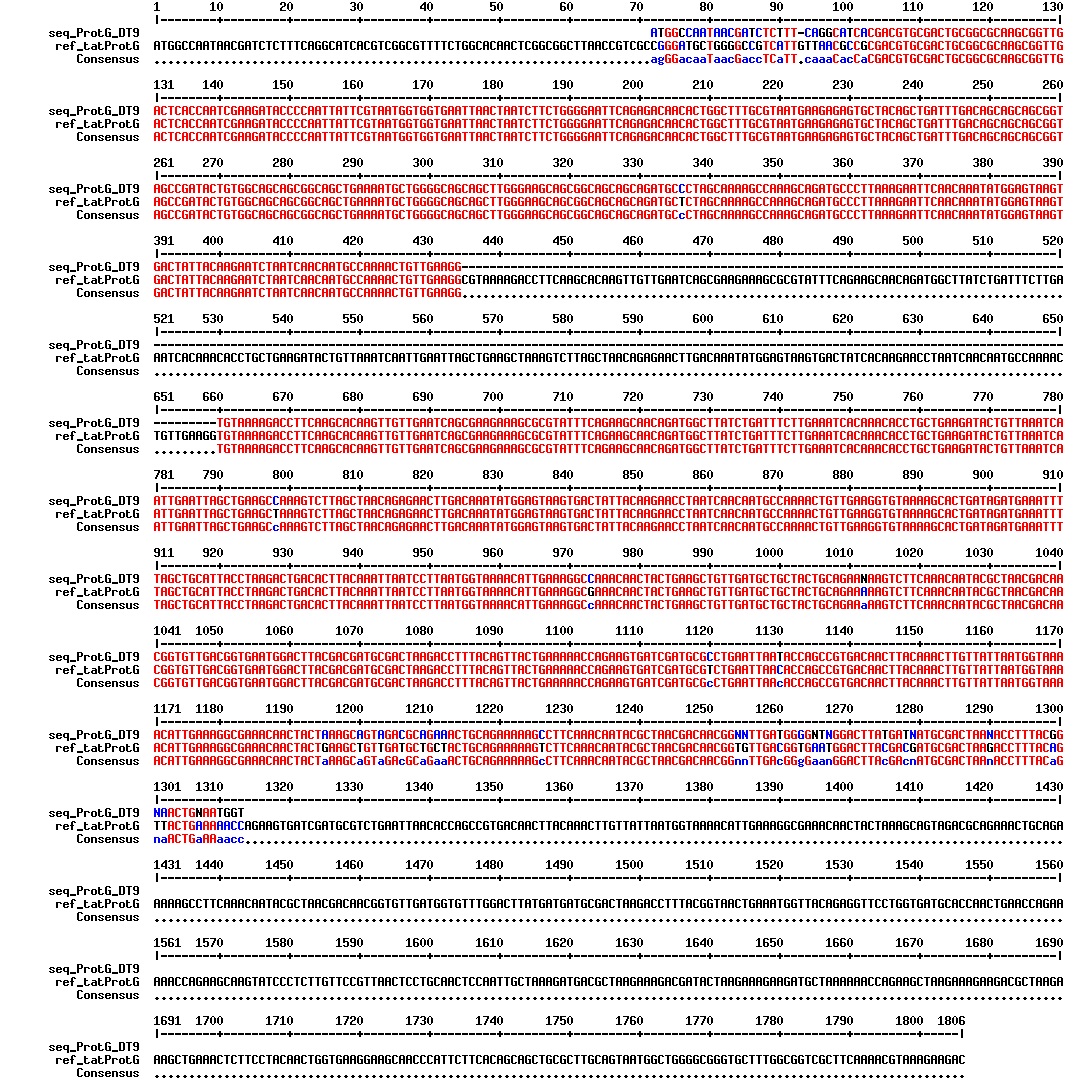 figures: Alignment of tat_ProteinG DT5, tat_ProteinG DT6 and tat_ProteinG DT9. ProteinG seems to be there but most of tat is absent. Only the first part of tat is there.
figures: Alignment of tat_ProteinG DT5, tat_ProteinG DT6 and tat_ProteinG DT9. ProteinG seems to be there but most of tat is absent. Only the first part of tat is there.
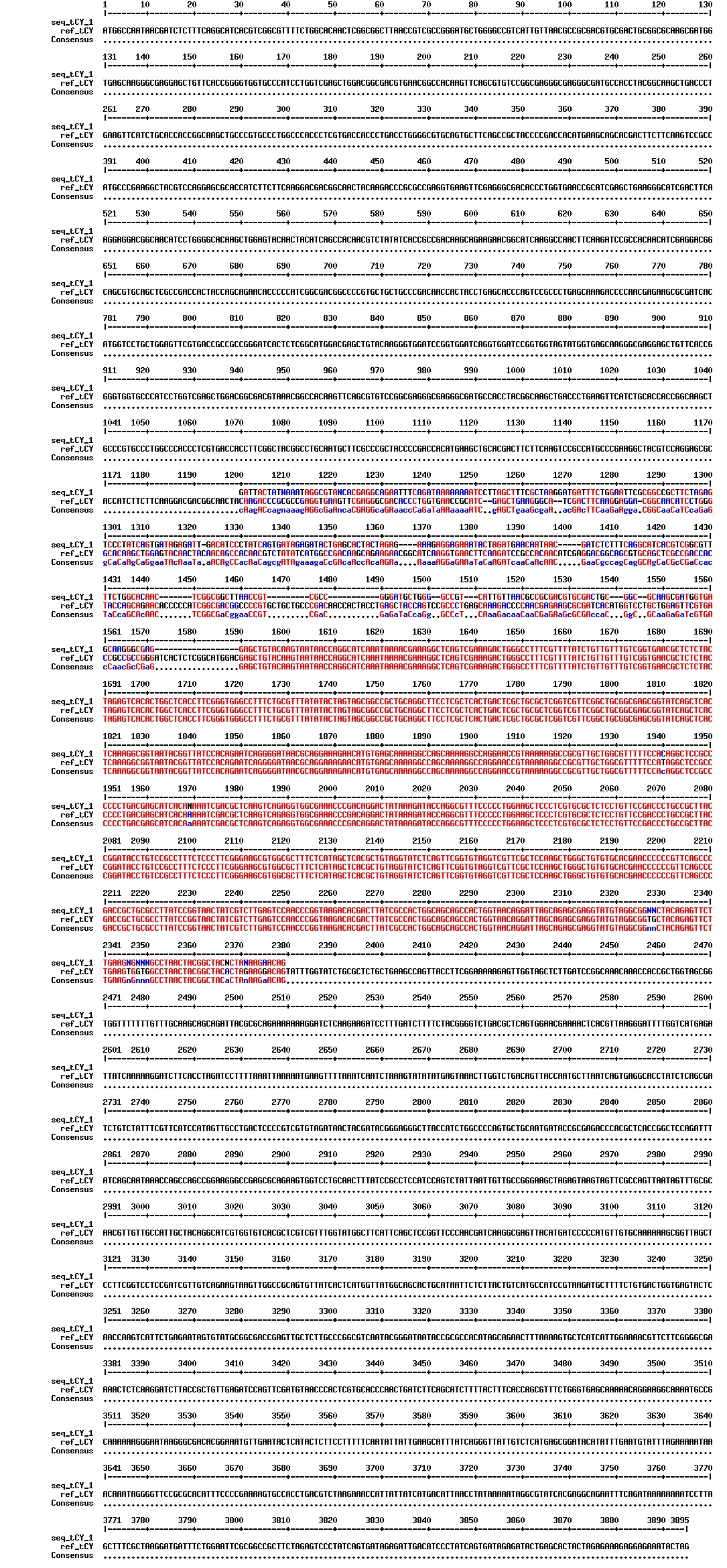
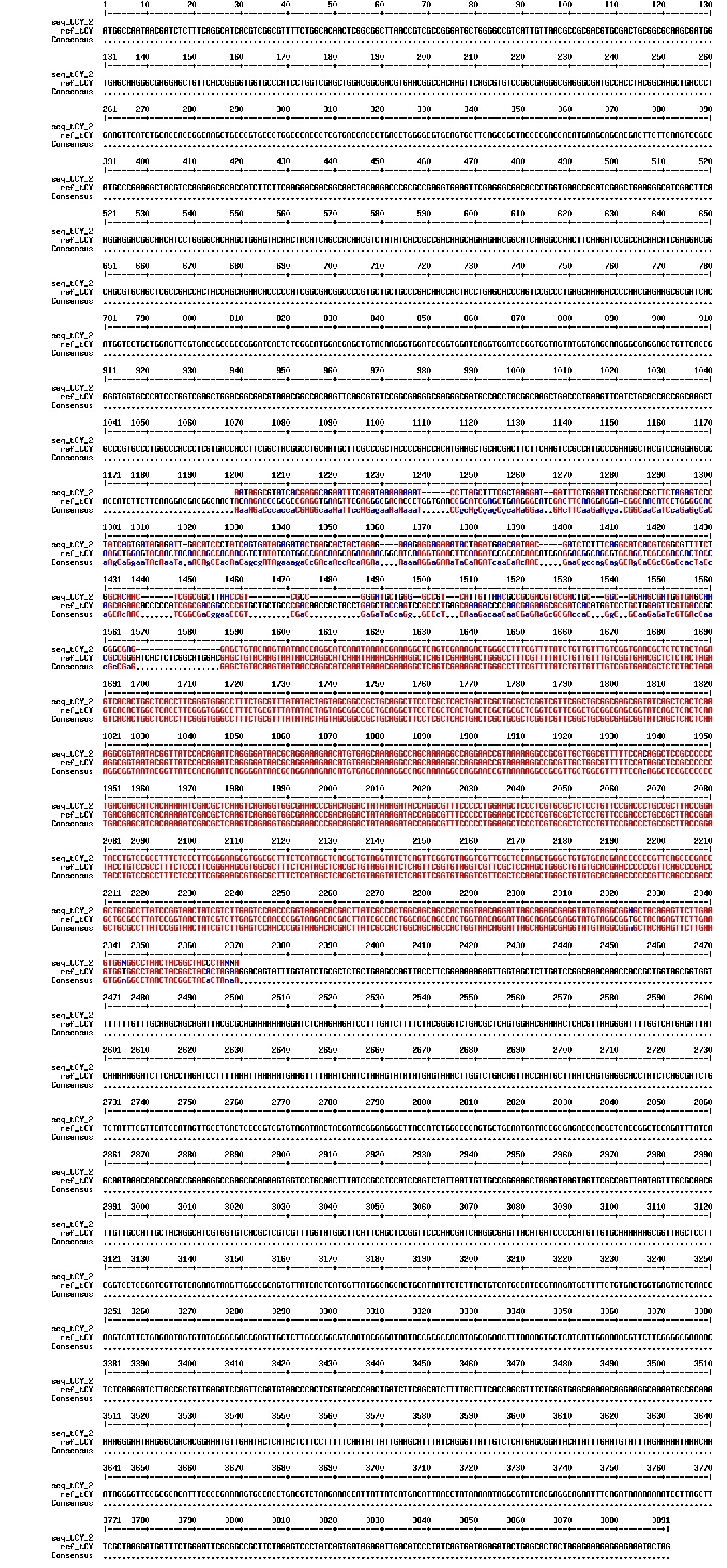
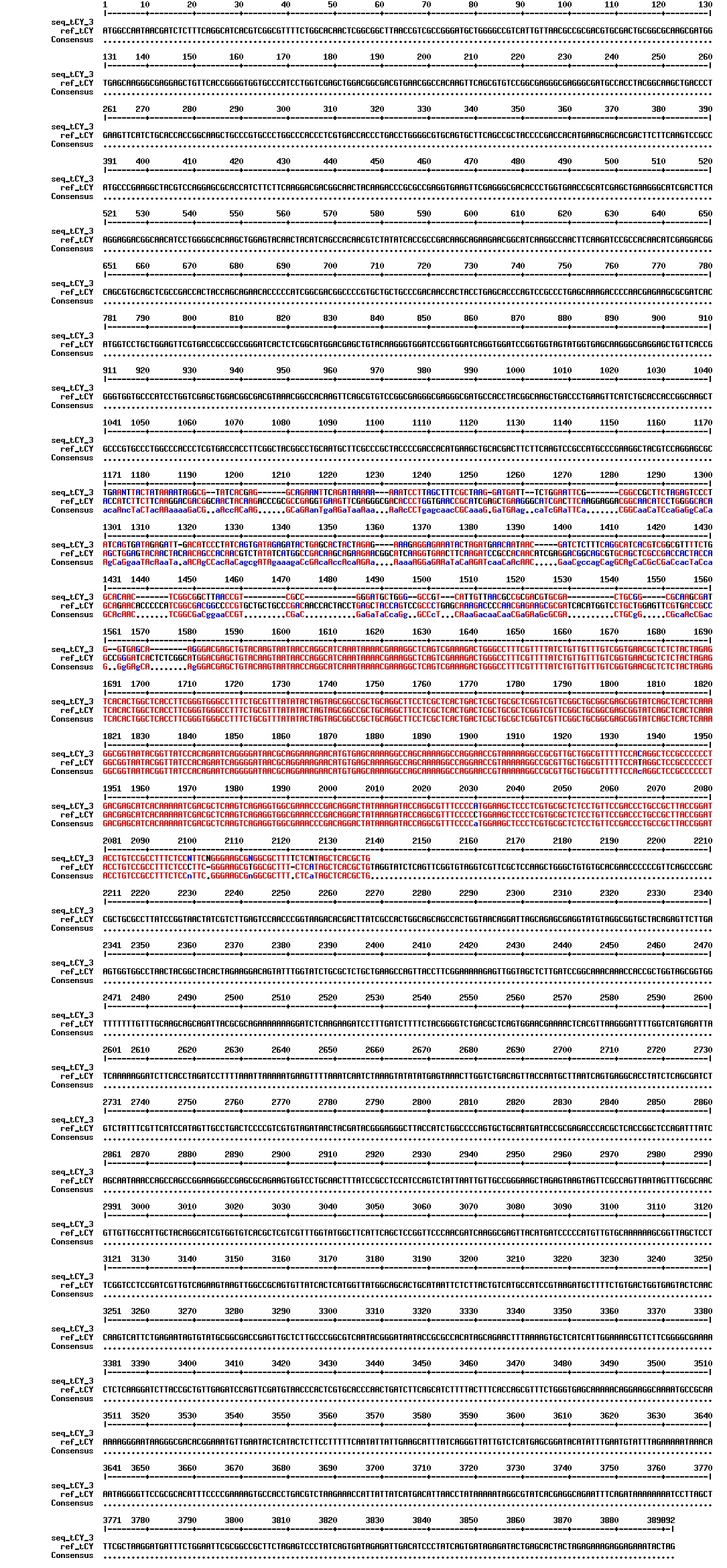
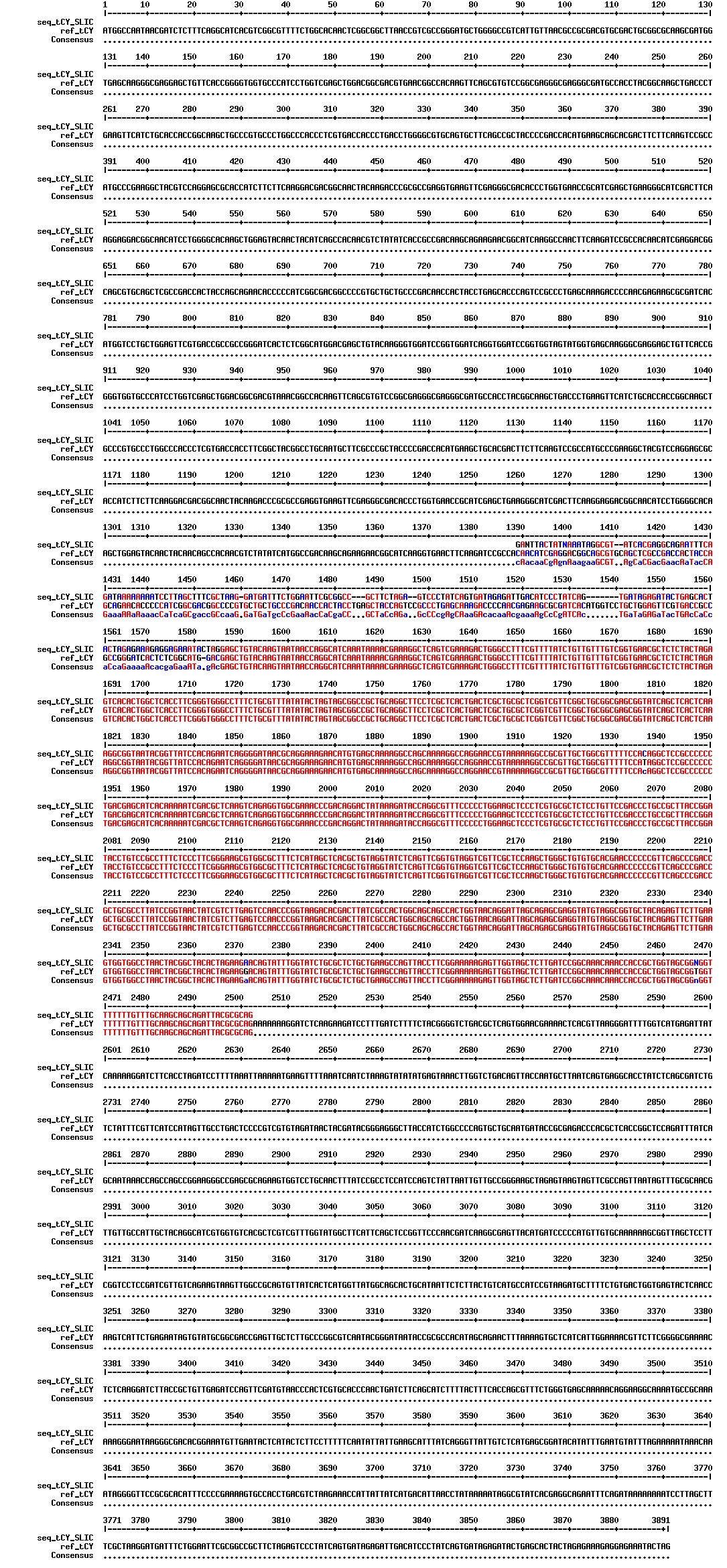 figures: Alignment of Gibson tCY 1, Gibson tCY 2, Gibson tCY 3 and SLIC tCY. tat and YFP is present, but CFP is absent in all of these sequences.
figures: Alignment of Gibson tCY 1, Gibson tCY 2, Gibson tCY 3 and SLIC tCY. tat and YFP is present, but CFP is absent in all of these sequences.
Thursday 05.09.13
We wanted to try to fix the addition and deletion in tat-ProteinG DT and S3 (respectively) by using the forward primer for tat (F_pl.b_tat) that has the right sequence. PCR-reactions for amplifying the backbones (BB) and the tat_ProteinG of both DT8 and S3 were run. These 4 samples were then run on a gel to confirm the results (figure below).
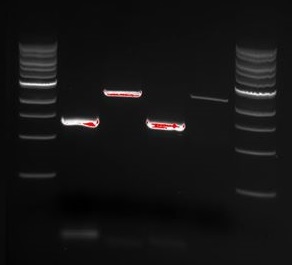 figure: PCR products of tat_ProteinG and BB from DT8 (well 1 and 2) and S3 (well 3 and 4). Ladder applied is [http://66.155.211.155/nebecomm/products_intl/productN3232.asp 1 kb DNA ladder]. The fragments nicely matches the expected sizes of 2800 bp for backbone and 1300-1400 for tat_proteinG.
figure: PCR products of tat_ProteinG and BB from DT8 (well 1 and 2) and S3 (well 3 and 4). Ladder applied is [http://66.155.211.155/nebecomm/products_intl/productN3232.asp 1 kb DNA ladder]. The fragments nicely matches the expected sizes of 2800 bp for backbone and 1300-1400 for tat_proteinG.
The PCR products were also incubated at 37 °C with Dpn1 to remove all old plasmids. The PCR products where then used in direct transformation of E.coli DH5α. The fragments that originated from the same plasmid where used together (BB from DT8 with tat_PrG from DT8 and so on). There was also performed a direct transformation tat_GFPmut3 with BB from S3. There were made two parallels of all combinations yielding 6 direct transformation. Controls with all the 5 fragments used was also prepared.
Friday 06.09.13
PCR
Turned the Pm XylS promoter system into a biobrick using PCR. The following recipe was used:
- 31.5 μl dH2O
- 10 μl 10x phusion buffer
- 1 μl dNTPs
- 2.5 μl fwd primer diluted 1:10 compared to stock solution (primer sequence: GAATTCGCGGCCGCTTCTAGTCAAGCCACTTCCTTTTTGC)
- 2.5 μl rev primer diluted 1:10 compared to stock solution (primer sequence: TACTAGTAGCGGCCGCTGCAGTGCATAAAGCCTAAGGGGTAGG)
- 1 μl template DNA
- 1 μl DMSO
- 0.5 μl Phusion DNA polymerase
Two different templates were used, and two samples were made for each template, giving in total four PCR batches. The first template was the Pm XylS plasmid, while the second was a PCR product of the Pm XylS promoter, modified in order to eliminate an XbaI restriction site.
The following PCR program was used:
| Program step
| Temperature
| Duration
|
| Heated lid
| 103°C
| -
|
| Initial denaturation
| 98°C
| 30 s
|
| Cycle 30x
|
|
|
| Denaturation
| 98°C
| 15 s
|
| Annealing
| 64°C
| 15 s
|
| Elongation
| 72°C
| 53 s
|
| End of cycle
|
|
|
| Final elongation
| 72°C
| 10 min
|
| Hold
| 4°C
| ∞
|
Gel picture of the PCR products:
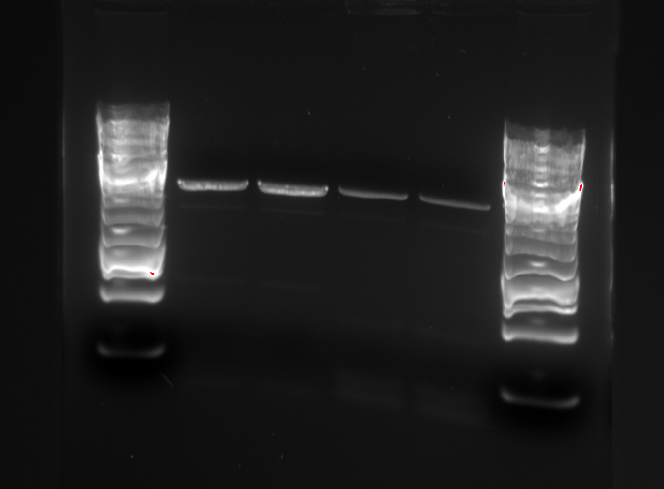
The PCR products were rinsed using the QIAgen PCR Purification kit. The two parallel samples using the same template were eluted in the same eppendorf tube. The following concentrations were measured:
| BioBrick
| Concentration
|
| Pm XylS from BB template
| 98.3 ng/μl
|
| Pm XylS with XbaI site
| 171.9 ng/μl
|
Miniprep
<partinfo>BBa_E0240</partinfo> and <partinfo>pSB3K3</partinfo> were isolated using the Wizard Plus SV Minipreps DNA Purification System kit. The following concentrations were measured after the isolation:
| Part
| Concentration
|
| <partinfo>BBa_E0240</partinfo>
| 79.8 ng/μl
|
| <partinfo>pSB3K3</partinfo>
| 42.8 ng/μl
|
Tuesday 10.09.13
Restriction digest
Linearized pSB1C3 and the two versions of Pm XylS (modified and unmodified, one of them containing an XbaI restriction site) were cut with EcoRI and PstI in order to put the promoter system in the shipping plasmid. The following volumes of DNA and water were used in the digestions:
| DNA part
| Concentration of DNA
| Calculated volume of DNA used in digestion
| Volume of dH2O used in digestion
|
| Linearized pSB1C3
| 25 ng/μl
| 10 μl
| 6 μl
|
| Pm XylS BB
| 98.3 ng/μl
| 2.5 μl
| 13.5 μl
|
| Pm XylS XbaI
| 171.9 ng/μl
| 1.5 μl
| 14.5 μl
|
Buffer 3 were used in all restriction digests. In the table above, Pm XylS BB refers to the biobrick that has been modified compared to the original Pm XylS promoter, which contains an illegal XbaI restriction site. Pm XylS XbaI is the original promoter, that contains the XbaI site.
After the restriction digest, the products were purified using the QIAquick PCR Purification Kit, which has a filter size that allows the short ends of the prefix and suffix of the linearized plasmids and biobricks that should be present after the restriction digest, to pass through the filter. The following concentrations were measured after the purification:
| DNA part
| Concentration
|
| pSB1C3
| 4.2 ng/μl
|
| Pm XylS BB
| 6.8 ng/μl
|
| Pm XylS XbaI
| 7.6 ng/μl
|
Gel electrophoresis
Gel electrophoresis were performed to ensure that the right DNA fragments are present after the restriction digest.

In this picture, the first band is pSB1C3, which linearized should be 2070 bp long. The Pm XylS promoter system has a length of 1759 bp, and the modified and unmodified version of the promoter system corresponds to band 2 and 3.
Ligation
pSB1C3 was ligated to Pm XylS BB and Pm XylS XbaI, respectively. The following ligation mix was set up. The same mix were used for both promoter systems, since the concentrations were almost the same. The volume of backbone used corresponds to 25 ng of DNA, which is the recommended amount of backbone suggested on the partsregistry website.
| Component
| Volume
|
| pSB1C3
| 6 μl
|
| Pm XylS BB / Pm XylS XbaI
| 7 μl
|
| T4 DNA ligase buffer
| 1.5 μl
|
| T4 DNA ligase
| 0.5 μl
|
The ligation mix was put in the fridge for overnight ligation.
 "
"






































