 "
"
Team:SYSU-China/Project/Modeling
From 2013.igem.org
| Line 481: | Line 481: | ||
<br/><img src=" https://static.igem.org/mediawiki/2013/c/c1/Modeling_21.png " width="300" /><br /> | <br/><img src=" https://static.igem.org/mediawiki/2013/c/c1/Modeling_21.png " width="300" /><br /> | ||
| - | |||
| - | |||
<h2> The excursion of little mathematician--Data analysis of the FACS data | <h2> The excursion of little mathematician--Data analysis of the FACS data | ||
</h2> | </h2> | ||
| Line 536: | Line 534: | ||
And, let’s wait for the result. | And, let’s wait for the result. | ||
</p> | </p> | ||
| + | |||
| + | <h2> | ||
6.reference | 6.reference | ||
Revision as of 22:18, 27 September 2013
ipsc

1.Overview
Modeling is a powerful tool in synthetic biology and engineering. In the iPSCs Safeguard project, modeling has provided us with an important engineering approach to characterize our pathways and predict their performance, thus helped us with modifying and testing our designing.
Basically, the models built by us can be divided into two levels. On cell level, we proposed the multi-compartment model to trace the change of the iPS cells in different time nodes, thus we are able to describe the growth and decay of iPSCs. The number of cells at the initial stage, growth rate and death rate of cells caused by suicide gene in our Safe-guard pathway were all taken into account. Dox and miRNA concentration acted as parameters switching the pathway state and affected the growth dynamics of iPSCs.
On gene level, we adopt a chemical kinetics modeling approach to analyze the interaction between transcriptional factor tTA, co-repressor Dox, target mRNA and miR122. Our analytical framework is based on the use of ordinary differential equations, we described the expression variation of target mRNA in different state of the pathway, determined by Dox and miR122 level. The models at gene and cell level worked in concerts to give us a better understanding of how our pathway and cell lines functions and how to improve the designing.
2. Multi-compartment model
2.1 Analysis of the problem
We firstly focus on factors that regulate the performance of the whole pathway. Protein tTA expressed by a EF1α promoter binds to the promoter pTRE to drive the transcription of target gene( in this case, eGFP or suicide gene) while Dox acts as a co-repressor prohibiting the transcription. MiR122 isa downstream part in the pathway after transcription of target mRNA, and mediated degradation of the mRNA, thus rescue the cell or knockdown its GFP expression. However, the miR122 level in iPSC was low and insufficient to exert obvious effect on the expression.
Apart from Dox concentration,we also monitored other parameters, including cell number after the stable infection and number of cell that survived the Suicide Gene. Moreover, we also kept track of fluoresence intensity of the control group who has been transfected with GFP, which can be employed to indicate the GOI expression level driven by Tet-Off system.
In pratical, we planned to monitor the cell group scale every 5 hours and technically, we counted the total clone area instead of cell number.
2.2 Symbols declaration and assumption
X1: initial number of iPS cells with Suicide Gene
X2: number of the iPS cells whose TRE have been combined with tTA
X3: number of iPS cells which have died from expressing Suicide Gene
k1: converting rate of the number of cells from phase X1 to phase X2
k2: converting rate of the number of cells from phase X2 to phase X3
The unit of ki(i=1,2) is hour-1.We measured it by dividing the absolute value of the cell number difference between former phase and latter phase, with the time period length.
Two cases are taken into account. In case (a), self-renewal and replication of cels are ingored while in case (b), we take that into consideration. To further simplify the model, we also assumed that every single cell in phase X1 turns into n1 state before phase X2, and every single cell in phase X2 turns into n2 state before phase X3. We simulated the kinetic process of gene expression and assumed an even distribution of cell content in the medium,after which the phase can be regarded as a compartment.
2.3 Solution
For each compartment, we construct unsteady state equilibrium equation, hence we obtain the ordinary equations

For case (b), we just need to modify the scalar coefficients of the equations above, and we obtain


We are going to solve X1(t), X2(t),X3(t), then we will plot the time course curve.
The initial conditions of the differential equations are as follows:
X1(0)= 5000 cells, X2(0)=0 cell, X3(0)=0 cell
k1=1day-1,k2=1 day-1
In case b, the replication time is set to be 26 hours and cells are considered to simply duplicate before switching into the next phase.Therefore, n1=n2=2. We use MATLAB ode45 function to solve the problem.
Source code
%igem_test1.m-Solution of the IPS cell differentiation model %using MATLAB function ode45.m to integrate the differential equations %that are contained in the file cell_diff_eq.m clc; clear all; %set the initial conditions, constants and time span xzero=[5000,0,0];tmax=4; k1=1; k2=1; tspan=0:0.1: tmax; N=3; %Integrate the equations [t X]=ode45(@cell_diff_eq,tspan,xzero,[],k1,k2); last=X(length(X),N); %Plot time curve plot(t,X(:,1),'-',t, X(:,2),'-',t, X(:,3),'-.'); legend('X1','X2','X3'); xlabel('time,days'); ylabel('number of cells'); function dx= cell_diff_eq(t,x,k1,k2) %cell expression kinetic procedure dx=[-k1*x(1); k1*x(1)-k2*x(2); k2*x(2); ];
The result of case (a):


3. Dosage effect of DOX in turning off the Tet-off system
DOX ,as is discussed above, hinders the binding of tTA to pTRE in Tet-Off system and knockdown expression of suicide gene. In our experiment, we employ fluorescence technique to manifest the amount of protein product by detecting the strength of the fluorescence.

TABLE 1. Experimental data

Our task is to find the proper curve to fit the sample data. First of all we plot the scatter diagram, and according to its tendency, we use type curve to fit the relation of GFP-DOX. We use MATLAB to aid our fitting, i.e. to determine the parameter a, b and k.

%expun.m function y=expun(s,t) %coefficient and variable y=s(1)+s(2)*exp(-s(3)*t) %curvefit.m treal=[0 0.125 0.25 0.5 1 2]; %experimental data yreal=[25 13 10 8 6 5.7]; s0=[0.2 0.05 0.05]; %iteration initial value sfit=lsqcurvefit('expun',s0,treal,yreal); %least square curve fit f=expun(sfit,treal); disp(sfit);
The result :

So a=6.4147,b=18.3999,k=7.3173.
Then we program the diagram file GFP-DOX.m
%GFP-DOX curve treal=[0 0.125 0.25 0.5 1 2]; %experimental data yreal=[25 13 10 8 6 5.7]; t=0:0.1:2.5; a=6.4147;b=18.3999;k=7.3173; y=a+b*exp(-k*t); plot(treal,yreal,'rx',t,y,'g'); xlabel('Dosage of DOX'); ylabel('GFP');

As is shown in the figure above, we can conclude that the amount of GFP tend to be steadily over 1.5 ug, the higher concentration of DOX we set, the lower GFP we expect. However, under the real experimental conditions, over 2.2 ug DOX will lead to the undesired necrosis of the cells. This is a trial-experiment which proved that such a balance point for good turning- off effect and cell tolerance does exist in a certain interval concentration. More accurate experiment should be conducted on stable-transfected iPSCs to find the best cultivating condition.
4. Knockdown efficiency interpolation
According to the experimental data, here we use interpolation technique to find the relationship between miRNA-122 concentration, the number of miR122 target sites and cell knockdown efficiency, which leads to a function with two variables. The knockdown efficiency is represented by GFP expression level which is actually the ratio of the amount of GFP and that of the parameter GAPDH. The knockdown efficiency then is


FIGURE 4. 2 target sites, gradient miRNA concentration

TABLE2. Experimental data of 2 target sites, gradient miRNA concentration

TABLE3. Experimental data of 0.75ug miRNA plasmid with gradient target sites
We use the data above to do the interpolation. We use the griddata function to implement the interpolation.
MATLAB codes: clear miRNA=[0 0.025 0.05 0.1 0.25 0.75 0.75 0.75]; site=[2 2 2 2 2 1 2 4]; KD=[0 29 43 55 64 55 39 32]; cx=0:0.01:0.75; cy=0:0.05:4; cz=griddata(miRNA,site,KD,cx,cy','cubic'); meshz(cx,cy,cz),rotate3d %shading flat xlabel('miRNA(plasmid ug)'),ylabel('Target Site'),zlabel('knockdown efficiency(%)');

5. miRNA-mediated regulatory model
For any miRNA-mRNA complex, there are two architectures to describe the action of miRNA in the regulatory network. Correspondingly, there are two chemical kinetic models introduced to describe the performance of miRNA in the circuits.
The transcription-degradation architecture is based on the assumption that the miRNA takes effect at the latter stage of transcription and mediates the degradation of the mRNA that is undergoing formalizing. In this case the translation inhibition is speculated not to occur simultaneously with the transcription degradation, mRNA's expression depends on upstream factors. In our circuit, the transcriptional repressor is DOX, we denote it R, and there's no activator, To model the action of downstream mRNA gene G, we implement the nonlinear chemical kinetic equation. Let's denote the expression levels of mRNA, protein, miRNA and the repressor respectively by g(t),p(t),m(t), R(t), then we define the individual impact of proteinic repressor R on the change rate of g(t) by REP(t),

Where BSR represents the number of the binding sites; u represents the affinity constants with mRNA.
The generation proportion of mRNA committed by the DNA templates can be defined as

Since there is no activator, so f(t)=REP(t). Let r1 be the degradation rate of mRNA, r2 the translation rate of mRNA, r3 the interaction rate between mRNA and miRNA. Taking an arbitrary small time interval into account, namely [t,t+dt], degradation and interaction cut down the amount of mRNA while the transcription increases the amount of mRNA, thus we obtain.

The second interaction architecture for miRNA-mRNA complex points out that the upstream miRNA inhibits the translation of the downstream mRNA gene, resulting in the repression of the expression of the protein generated by mRNA gene. Translation inhibition architecture focuses on the change of the amount of the protein. A set of n upstream miRNAs denoted by m1,m2,m3,…,mn. We denote the concentration at time t of protein, mRNA, m1,m2,m3, …,mn by p(t), g(t), m1(t),m2(t),m3(t),…,mn(t). For mi,the repressive impact on translation is defined as

The comprehensive impact then is

Let r1 and r2 be the protein degradation rate and translation rate respectively. Degradation leads to the reduce of protein whereas the mRNA translation contributes to the increase of protein, so the ordinary differential equation is as follow

The excursion of little mathematician--Data analysis of the FACS data
When our project was proceeding, we found out an interesting problem, that is, how to calculate the kill efficiency of each suicide gene? And this quantity is also an important part of our modeling. Try to solve this problem,one of our mathematician, young Yang ZiYi, excursed a little bit from modeling to data analysis. And he analyzed our data of FACS.
One important point in mathematical analysis about the complicated biological system is, not to draw arbitrary assumption, arbitrary assumption just lead to disaster. Another important point is not to draw complicated assumption, which is hard to calculate. Base on these rules, Young ZiYi draw some simple and reasonable assumptions:
1. The initial condition of each parallel well is the same, that means, every well before transient transfection should have the same density, and the cells condition should be approximately the same.
2. In the GFP control group, the cells should be regarded as the same, whether they are transfected with GFP or not, since GFP do not harm the cells. But lipo-2000 will harm the cells, and may have some long term effect[1] so the GFP control group would be relatively weaker compare to negative control, which without any transfection manipulation;
3. The cells transfected with GFP should have an innate death rate r1 after 3 days cultivation. Besides, the ratio of “the initial number of cells” to “the number of cells harvested via FACS after 3 days” Should be the same, since if you sample any part of the wells you will observe the same distribution of cells, this means the cell experiment is scalable. Hence, we can define a special “state” to represent GFP cells in certain well, (r1,n1), n1 is the number of the cells we harvest in 30s, depends on r1 and the initial cell number.
4. In the well we transfect suicide genes, the cells which are transfected with suicide gene will be different from the cells which are not, due to the killing effect of suicide gene, and the cells without transfection will be at the same condition as GFP control and have the same r1. Cells transfected with suicide gene will have a different r2, we denote this part of cells by (r2,n2). If the transfection efficiency is a, then for any initial cell number N0, there will be N0×a cells transfected with suicide gene, the remaining N0×(1-a)are not. Here a in an unknown factor.
Then Young ZiYi built a model:
In the wells that had been tranfected with suicide genes, the final observed “state” (X, C) of the cells is an combination of two kinds of cells, one kind with state (r1,n1), the other kind with state (r2,n2). The effect of these two kinds will be summed up together. Then we can write down a function:
Because we can not easily get the transfection efficiency, we select a reasonable range from experience:[30%,60%], and solve the equation with the data from FACS, and get the following results:
1. In the GFP control group, the cells should be regarded as the same, whether they are transfected with GFP or not, since GFP do not harm the cells. But lipo-2000 will harm the cells, and may have some long term effect[1] so the GFP control group would be relatively weaker compare to negative control, which without any transfection manipulation;
1. In the GFP control group, the cells should be regarded as the same, whether they are transfected with GFP or not, since GFP do not harm the cells. But lipo-2000 will harm the cells, and may have some long term effect[1] so the GFP control group would be relatively weaker compare to negative control, which without any transfection manipulation;
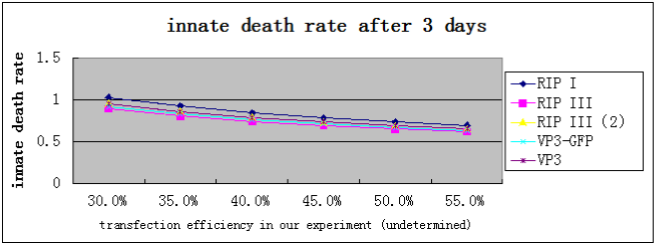
It turns out that, the death rate of cells transfected with suicide gene will be greater than 60%, significantly higher than normal cells and GFP control cells(29.07%, according to our data).
Then, We pick up one suicide gene, VP3, and take an “a” value 35% which we believe is fairly closed to the real tranfection efficiency, solve the equation and get the result r2 and n2. And then, we substitute the calculated r2 and n2 to the left side of the equation, and raise the value of “a” to predict what the death rate and the “number per 30 seconds” would be when the transfection efficiency is raised. The result is shown in a chart below
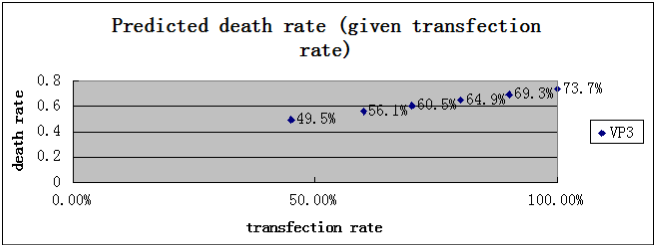

That makes our model become a real theory, because it predicts something that can be disproved. In the following day, we will try to raise our transfection efficiency, and try to comparing the result with the prediction.
And, let’s wait for the result.
6.reference
[1] Systems biology in practice concepts, implementation and application / (德) E. Klipp等著 ; 主译:贺福初, 杨 芃原, 朱云平 ,上海 : 复旦大学出版社, 2007
[2]Numerical methods in biomedical engineering / (美) Stanley M. Dunn, Alkis Constantinides, Prabhas V. Moghe著 ; 封洲燕译,北京 : 机械工业出版社, 2009
[3]miRNA regulatory circuits in ES cells differentiation: chemical kinetics modeling approach , Luo Z, Xu X, Gu P, Lonard D, Gunaratne PH, et al. (2011)
[4]kinetic signatures of microRNA modes of action, N Morozova, A Zinovyev, N Nonne, LL Pritchard - RNA, 2012
Sun Yat-Sen University, Guangzhou, China
Address: 135# Xingang Rd.(W.), Haizhu Guangzhou, P.R.China