 "
"
Team:Berkeley/Methods
From 2013.igem.org
| Line 140: | Line 140: | ||
<p>We followed the protocol from <a href=” http://www.bio-rad.com/webroot/web/pdf/lsr/literature/4110065A.pdf” _target=”new”>Bio-Rad’s Bradford Assay</a> to determine the concentration of purified FMO and GLU.</p> | <p>We followed the protocol from <a href=” http://www.bio-rad.com/webroot/web/pdf/lsr/literature/4110065A.pdf” _target=”new”>Bio-Rad’s Bradford Assay</a> to determine the concentration of purified FMO and GLU.</p> | ||
| - | <div style=" | + | <div style="width:80%;" align="center;"> |
<img src="https://static.igem.org/mediawiki/2013/c/c7/Bradford_Assay.jpg"> | <img src="https://static.igem.org/mediawiki/2013/c/c7/Bradford_Assay.jpg"> | ||
</div> | </div> | ||
| Line 156: | Line 156: | ||
</p> | </p> | ||
| - | < | + | <p>1. <b>FMO:</b> In each well of the TECAN plate, we added 0.3 mM of NADPH, purified FMO and various indole concentrations (0, 0.35, 0.50, 0.71, 1.02, 1.46, 2.09, 2.99, 4.27 mM) at 5% DMSO. </p> |
| - | + | ||
<p>Note: The 5% DMSO originates from the fact that indole had to be dissolved in DMSO. We recommend minimal DMSO because FMO is sensitive to DMSO.</p> | <p>Note: The 5% DMSO originates from the fact that indole had to be dissolved in DMSO. We recommend minimal DMSO because FMO is sensitive to DMSO.</p> | ||
| - | < | + | <p>1. <b>GLU:</b> In each well of the TECAN plate, we added purified GLU and various indican concentrations (0, 0.10, 0.19, 0.39, 0.78, 1.55, 3.10, 6.20, 12.40 mM) dissolved in water.</p> |
| - | < | + | <p>2. Using a TECAN plate reader, we determined the absorbance at 620 nm (peak wavelength at which indigo absorbs) every minute for 50 minutes. </p> |
| - | < | + | <p>3. The absorbance data was converted into concentration of indigo based on the indigo standard we have determined previously. </p> |
| - | < | + | <p>4. We wrote the following MATLAB code to automate analysis and generated the Michaelis-Menten Km.</p> |
| - | + | ||
<br></div> | <br></div> | ||
Revision as of 23:02, 27 October 2013

























Purpose:
When a particular protein sequence is known, but the codon usage per amino acid is not, designing primers that account for the codon degeneracy allows for PCR extraction of the given gene from a DNA template. This technique is especially useful when searching for a gene that has highly conserved regions in the amino acid sequence, such as actin, but may have different codon usage across the array of species in which the protein is found.
In iGEM 2013 Berkeley’s project, identifying a B-glucosyltransferase that acts on indoxyl was key, as a sequence for an indoxyl-specific B-glucosyltransferase does not exist in literature. Furthermore, none of the genomes of the indigo producing plant species have been sequenced, making our task even more difficult. By taking advantage of conservation patterns among B-glucosyltransferases from sequenced plant species related to indigo producing plants, we were able to design degenerate primers aimed to enrich cDNA libraries of the indigo plants for B-glucosyltransferases.
Materials:
- 1. DNA sequence editor (ApE)
- 2. Protein database (UniProt, NCBI)
- 3. BLASTX, PBLAST
- 4. Access to an Oligo synthesis facility
- 5. Sequence alignment software (Clustal Omega)
- 6. JalView 2.0 or other multiple sequences alignment viewer
- 7. Primer design software to ensure stability of designed primers
Method:
1. Identify gene of interest• In our case, we did not have access to the sequence of our protein of interest. So we relied on the information available for the general family of B-glucosyltransferases into which our desired enzyme falls.
2. Gather protein sequences of homologous genes to gene of interest• We first identified the taxonomic group that includes sequenced plant species and our indigo producing plants. This taxonomic group was the “core eudicotylions.” We then identified B-glucosyltransferases from sequenced plant species that have been characterized to act on substrates similar to indoxyl, and entered these protein sequences into the BLAST local alignment tool. This tool allows users to gather other related proteins with high sequence similarity to the query. After performing several iterations of searches like these using a variety of B-glucosyltransferases, we assembled our 200 sequences into a single document in FASTA format.
3. Generate multiple sequence alignment (MSA) of gathered homologs• Using a downloadable alignment tool such as MAFFT or ClustalΩ allows the user to generate an MSA from the gathered sequences. An MSA indicates the degree of conservation among the selected homologs, with dashes showing no conservation.
4. Use MSA editor/viewer to visualize and interpret MSA• Jalview 2.0 was used by our team to visualize the B-glucosyltransferase MSA. To easily identify conserved regions, a coloring scheme based on percent identity was applied, which showed highly conserved regions in dark blue. In the set of B-glucosyltransferases we gathered, a highly conserved region of 7 amino acids was identified. Further research into this region showed that it is referred to as the PSPG box, a sequence of amino acids that allow the enzyme to recognize UDP-glucose, the donor substrate in our pathway. We decided to take advantage of this PSPG box and a similarly conserved region towards the beginning of the MSA to enrich cDNA libraries generated from a variety of indigo producing plants for B-glucosyltranferases.

• For the PSPG box, the conserved amino acids we decided to take advantage of were HCGWNS. Each amino acid has a different number of codons that can code it. For example, W, tryptophan, only has a single codon that codes for it while S, serine, has 6 codons that code for it. Serine can be coded by either an A or T in the first codon position, a C or G in the second, and either A, C, G, or T in the third. This gives serine a total degeneracy of 16, indicating that there are 16 combinations of codons that must be generated in the primers to account for all different nucleotide combinations that can code serine. A simple way to represent the degenerate sequence for the serine codon is by writing WSN, where each letter corresponds to some subset of nucleotides. A complete list of degeneracies per amino acids and the corresponding single letter code for nucleotides is given.
• Choose the appropriate degenerate sequences for each amino acid that is conserved. Assemble these together and generate your degenerate primer! Ensure that the melting temperatures of the forward and reverse primers to be used in a single PCR are similar. The final primers we designed for the PSPG box are shown below.

• To ensure that the overall concentration of primers is sufficiently high, primers with 128-fold degeneracy or less are recommended by Springer protocols. The complete degeneracy of the primer is obtained by multiplying together the degeneracy of each amino acid in the designed sequence.
• The primer should be 20-23 nucleotides long to ensure annealing.
• The gene of interest to be extracted should be between 200-1000 bps long to ensure that short PCR fragments do not dominate.
• If degeneracy is too high, using multiple sets of primers that have slightly less degeneracy could provide better results.
• If using a eukaryotic cDNA library as a template for PCR, taking advantage of the 5’ poly-T tail for PCR may prove useful. As long as a fraction of the gene is extracted, sequencing and further primer design could eventually extract the full gene. If using a 5’ RNA addition while generating a cDNA library, oligos for the 5’ addition could also be used for PCR.
• Using inosine (I) as a substitute for N or H nucleotides could reduce degeneracy between 2-4 fold.

Purpose:
Due to the instability of RNA, generation of cDNA is ideal for the study of your transcriptome of interest. The type of cDNA library will be dependent on the downstream applications that you need. If you know the DNA sequence of your gene of interest, then a simple reverse transcription reaction will suffice. Reverse transcription relies on the existence of the Poly(A) tail of eukaryotic mRNA. Similar to PCR, reverse transcription uses a primer that binds the Poly(A), and a polymerase enzyme.

If you don’t know the DNA sequence of your coding region, then a known sequence at the 5’end is necessary for full amplification. For the BlueGenes iGEM project, we are searching for a glucosyltransferase of unknown sequence. Here, we describe the generation of a cDNA library for the purpose of screening it with degenerate primers.
Materials:
- 1. mRNA extracted from plant tissue (Concentration above 500ug/ul is ideal)
- 2. GeneRacerTM Kit (Life Technologies)
- 3. SuperScript III RT module – GeneRacerTM
- 4. Mini centrifuge with temperature control (4C)
Since RNA is the least stable of the nucleic acids, it is important to be particularly careful while following this procedure. Your best friends when working with RNA will be temperature and time. To prevent degradation, it is important that your sample remains cold, and that you work fast. In addition, maintain general sterile technique and use RNAse free materials.
Methods:
GeneRacerTM contains protocols to dephosphorylate RNA, de-cap RNA, and ligate an RNA oligo to the 5’ end. We deviated only slightly from the protocols published by Life Technologies.
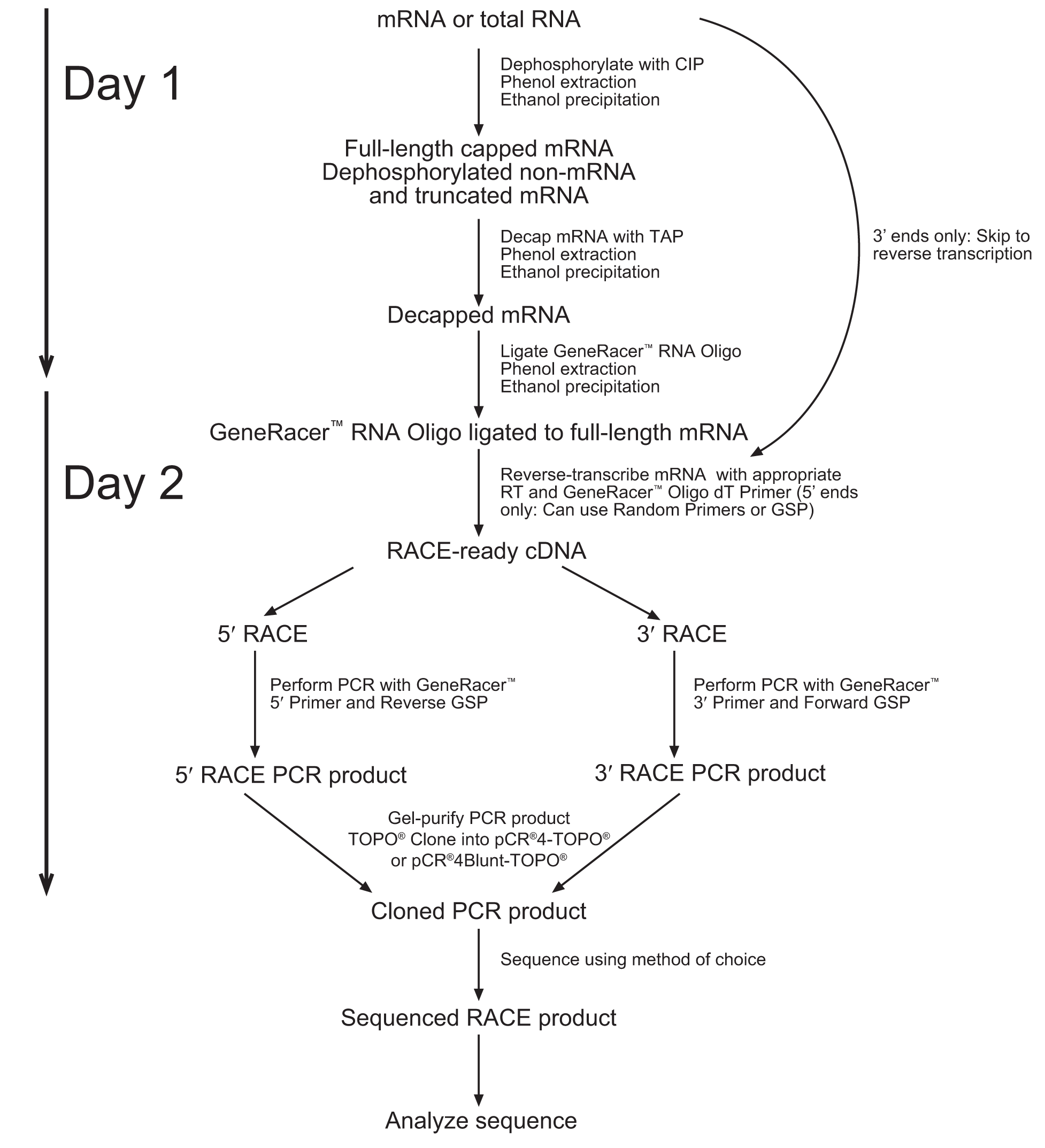
1. Note that the diagram below suggests a 2 Day process. We obtained better results when following the entire protocol without stopping (Unfortunately, this resulted in spending the night in the laboratory). The entire process takes about 10-12 hours from RNA extraction to cDNA.
2. Due to the lack of sequence data for our coding region. We followed the 5’Race protocol (which includes the addition of an RNA oligo to the 5’ end), but performed a reverse transcription reaction with the use of the Poly(A) binding GeneRacer primer.
1. Controlling for genomic contamination can be carried out by simple PCR of a known gene that contains introns. The difference in size of gel bands will demonstrate if amplification happened from a mature mRNA or some other nucleic acid contamination.
2. The kit contains HeLa mRNA, which serves as a control for the successful generation of cDNA. Note that their actin primers are generally good to test other tissues, but creating your own control primers (from a known gene of the organism) provides an added degree of confidence.
3. cDNA will usually be too concentrated at the end of the protocol. Check the specifications of your favorite PCR protocols for ideal concentrations.
4. Remember that cDNA is a single stranded nucleic acid, which can form stable secondary structures. Use of DMSO, and hot-start PCR generally gave us better amplification results.
Purpose:
The Berkeley iGEM team 2013 has determined the enzyme kinetics of FMO and GLU (data for: FMO and GLU) based on the Michaelis-Menten kinetics model. Our purpose of determining kinetics was to give a quantitative data of how fast these enzymes can turnover their respective substrates, which is very useful when considering scaling up the process and for future iGEM teams when they use our enzymes.
Materials:
- Purified Enzyme: FMO or GLU
-
- Substrate:
- FMO: Indole, NADPH (co-factor)
- GLU: Indican
- TECAN Plate Reader and TECAN Plate
- Solvents: DMSO, Water
Methods:
1. Determining the Concentration of Purified EnzymeWe followed the protocol from Bio-Rad’s Bradford Assay to determine the concentration of purified FMO and GLU.

We determined the concentration of our purified enzyme to be 4.2 mg/L for FMO and 3.5 mg/L for GLU.
2. Determining the Michaelis Menten kinetics
IMPORTANT: We assumed that the oxidation step from indoxyl to indigo is much faster than the formation of indoxyl via FMO (indole to indoxyl) or GLU (indican to indoxyl).
1. FMO: In each well of the TECAN plate, we added 0.3 mM of NADPH, purified FMO and various indole concentrations (0, 0.35, 0.50, 0.71, 1.02, 1.46, 2.09, 2.99, 4.27 mM) at 5% DMSO.
Note: The 5% DMSO originates from the fact that indole had to be dissolved in DMSO. We recommend minimal DMSO because FMO is sensitive to DMSO.
1. GLU: In each well of the TECAN plate, we added purified GLU and various indican concentrations (0, 0.10, 0.19, 0.39, 0.78, 1.55, 3.10, 6.20, 12.40 mM) dissolved in water.
2. Using a TECAN plate reader, we determined the absorbance at 620 nm (peak wavelength at which indigo absorbs) every minute for 50 minutes.
3. The absorbance data was converted into concentration of indigo based on the indigo standard we have determined previously.
4. We wrote the following MATLAB code to automate analysis and generated the Michaelis-Menten Km.