 "
"
Team:NTNU-Trondheim/Project
From 2013.igem.org
Gram negative bacteria export numerous proteins into the periplasm. The twin-arginine translocation pathway
(Tat pathway) is a protein export, or secretion pathway found in plants, bacteria, and archaea. In contrast to
the Sec pathway which transports proteins in an unfolded manner, the Tat pathway serves to actively translocate
folded proteins across a lipid membrane bilayer. By using this transportationsystem we aim to introduce new proteins into the periplasm of bacteria. Once in the periplasm the protein will to some extent end up in outer membrane vesicles (OMV's) that budd of the bacteria.
As all gram negative bacteria produce outer membrane vesicles, we looked into the content of these vesicles.
Was the sorting of proteins random? Could we direct certain proteins toward them? And what function would that
give? Can we use this to our advantage?
Studies show that a twin-arginine signal peptide is able to direct the export of active green fluorescent
protein (GFP) in E.coli and that translocation almost exclusively occur by the Tat-pathway. With this in mind
we proceeded with making a construct containing tat and a GFP. As GFP's are poplular reportergenes we thought it
would be fun to explore them further. As the absorbtionspectrums of different GFP's are quite similar we wondered
how they might look when dimerized. As part of our project we will design a fluorescent protein dimer with GFP and
red fluorescent protein (RFP). In addition we will fuse them with the tat signal peptide that will direct them
into the periplasm and further to the OMV's. The FP-dimer construct will be a BioBrick. As the goal of the project is to determine if vesicles can be utilized for drug delivery we want to see if they can be masked from the immunesystem by introducing a specific protein.
Protein G is known to bind to Human Serum Albumin which helps S.dysgalactiae subsp. equisimilis hide
from the immune system. Protein G could therefore be a potential important piece in a drug carrier by masking it
from immunological destruction. Introducing protein G into vesicles also demonstrate that it is indeed possible
to manipulate the content and therefore the properties of OMV's.
As our constructs will be put into plasmids for cloning we decided to make a new promoter BioBrick.
Gram negative (and some gram positive) bacteria produce outer membrane vesicles (OMV) by outward bulging of the outer plasma membrane. These OMV has several properties such as quorum sensing, biofilm development, involvement in pathogenesis (some for pathogenic bacteria), DNA transfer and transport of enzymes to distal locations [1]. The OMVs contain different proteins on their membrane surface and within their membrane with the most abundant proteins being OmpF, OmpC and OmpA [2]. When running a SDS-PAGE on a vesicle sample a protein distinct pattern will arise as illustrated in the figure below:

Table: A typical SDS-PAGE of a purified vesicle sample.
The ability of some pathogenic bacteria to use their OMVs in pathogenesis is particularly interesting. Pseudomonas Aeruginosa and pathogenic E.coli has been studied to some extent for their OMVs role in pathogenesis. These vesicles contains virulence factors such as proteases, pro-inflammatory proteins and toxins, and attack eukaryotic cells by binding on the surface and then internalize (see figure below) [3].

Table: Gold labeled vesicles from enterogenic E.coli that binds and internalizes in HT29 cancer cells [3].
The vesicles will display a lot of the same proteins as the bacteria as they bulge of from the outer membrane. This trait, and the fact that the vesicles will not be able to multiply on their own, has qualified them as a possible candidate for vaccines. Engineered vesicles has been shown to be immunogenic in mice, but more research remain before OMV as vaccines can be tested on humans [4]].
Whereas OMVs function and contents has been studied for decades, their potential as a drug carrier has not previously been investigated. As it is still many obstacles for an effective production method of different drug carriers within the field of nano-medicine, exploiting gram negative bacteria OMV production could be a possibility for drug delivery in the future. Making drug carriers synthetically in small sizes is difficult and the vesicles measure only 20-200 nm in size which makes them attractive for this purpose. Especially vesicles from pathogenic bacteria could be further manipulated as these vesicles naturally can attack human eukaryotic cells. The issue is that they contain PAMP's (Pathogen Associated Molecular Patterns) that are recognized by the immune system. Gram negative bacteria contain LPS (Lipopolysaccharide) in their outer membrane that is recognized as foreign by immune cells. To stop the immune cells from attacking the vesicles we propose to introduce the outer membrane protein,Protein G, into the vesicles. This protein binds Human Serum Albumin (HSA) which tricks the immune system into thinking the vesicles are harmless blood cells. This will make the vesicles more suitable as a drug delivery vehicle.

Figure: Immune cells recognize and attack vesicles.

Figure: Protein G binds Human Serum Albumin and masks the vesicles from the immune system.
The first fluorescent protein (FP) to be discovered was the green fluorescent protein (GFP) isolated from Aequorea victoria. Many other different fluorescent protein (FP) with different colours (red, cyan, yellow and other version of these) has since been created by mutating GFP. The FPs has been crucial in terms of visualising cells and determining intracellular proteins by fusing the FPs to proteins of interest. Having a variety of FPs gives scientists the opportunity to study different proteins at the same time.
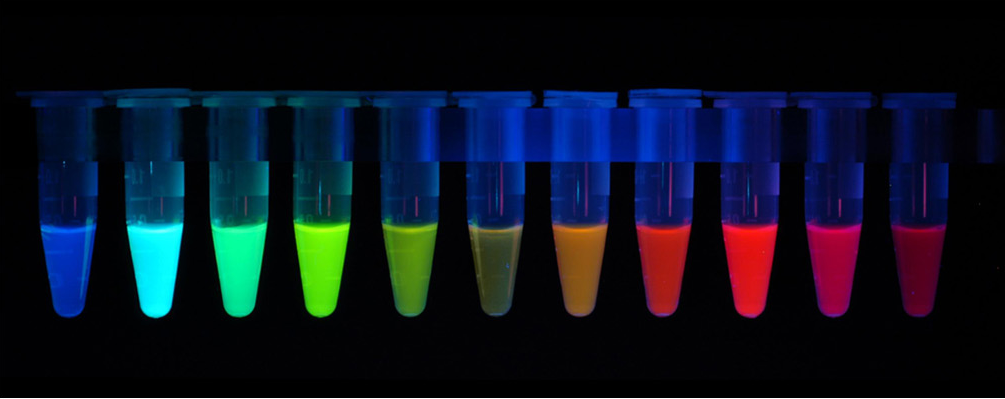
Figure: Fluorescent Proteins.
The discovery of green fluorescent protein in the early 1960s eventually created a new era in cell biology by assisting investigators to apply molecular cloning methods, fusing the fluorophore moiety to a wide variety of protein and enzyme targets, in order to monitor cellular processes in living systems using optical microscopy and related methodology[5].
Fluorescent proteins are structurally homologous proteins that share the unique property of being self-sufficient to form a visible wavelength chromophore from a sequence of 3 amino acids within their own polypeptide sequence. Biologists introduce a gene (or a gene chimera) encoding an engineered fluorescent protein into living cells and subsequently visualize the location and dynamics of the gene product using fluorescence microscopy.
The presence of a fluorescent component in the bioluminescent organs of Aequorea victoria jellyfish was introduced by Davenport and Nicol in 1955, but it was Osamu Shimomura who was the first to recognize that this fluorophore is actually a protein. He mentioned in one of his papers A protein giving solutions that look slightly greenish in sunlight though only yellowish under tungsten lights, and exhibiting a very bright, greenish fluorescence in the ultraviolet of a Mineralite that has been isolated from squeezate. Squeezate describing a solution that resulted from the squeezing of the excised bioluminescent tissues of the jellyfish through a cotton bag. After that Shimomura reported the fluorescence emission spectrum of this protein and also he suggested that energy transfer from aequorin to this green fluorescent protein could explain why the in vivo luminescence of Aequorea is greenish and not blue like the luminescence of purified aequorin[6].
The gene for green fluorescent protein was first cloned in 1992, but the important possibility to be used as a molecular probe was not achieved until several years later when fusion products were used to track gene expression in bacteria and nematodes. Green fluorescent protein has been engineered to produce numbers of various colored mutants, fusion proteins, and biosensors which are broadly referred to as fluorescent proteins. Recently, fluorescent proteins from other species are also identified and isolated, resulting in further expansion of the color palette. With the rapid evolution of fluorescent protein technology, the utility of this genetically encoded fluorophore for a wide spectrum of applications beyond the simple tracking of tagged biomolecules in living cells is now becoming fully appreciated.
A broad range of fluorescent protein genetic variants have been developed that feature fluorescence emission spectral profiles spanning almost the entire visible light spectrum. Mutagenesis efforts in the original Aequorea victoria jellyfish green fluorescent protein have resulted in new fluorescent probes that range in color from blue to yellow, and are some of the most widely used in vivo reporter molecules in biological research. Longer wavelength fluorescent proteins, emitting in the orange and red spectral regions, have been developed from the marine anemone, Discosoma striata, and reef corals belonging to the class Anthozoa. Still other species have been mined to produce similar proteins having cyan, green, yellow, orange, and deep red fluorescence emission. Developmental research efforts are ongoing to improve the brightness and stability of fluorescent proteins, thus improving their overall usefulness[7].

Figure: Fluorescent Protein Excitation and Emission Spectra.
Fluorescence is basically a color-resolved technique, to choose a right FP is important to consider its spectral profile, that is, the color of its fluorescence. A broad range of FP variants that span nearly the entire visible spectrum has been developed and optimized. For comparing the brightness of various FPs, each normalized excitation spectrum is multiplied by its peak molar extinction coefficient, and then divided by the peak molar extinction coefficient of EGFP. Likewise, each normalized emission spectrum is multiplied by its molecular brightness (molar extinction coefficient × quantum yield) and then divided by the brightness of EGFP. [8].
Choosing of the best-performing FPs in each color class are based on a number of critical factors, including maturation efficiency, spectral properties, photostability, monomeric character, brightness, fidelity in fusions and potential efficiency as a Förster resonance energy transfer (FRET) donor or acceptor.[9].
The blue, green, and yellow FPs appear to have reached their potential, but improvements are likely to continue for orange, red and combinations of different FPs. Each FP has its own defined emission and excitation spectrum.

Table: Fluorescent Protein Excitation and Emission Wavelength.
By using multi-color fluorescence microscopy, FPs are often used in combination to examine interactions between their fusion partners. In the second part of our project, we want to create new combinations of FPs. We intend to make this happen by fusing different FPs together into a new construct, namely GFP, red fluorescent protein (RFG), cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP). This will give us new combinations of FPs with new absorption spectrum. The goal of this work was to expand the color palette of FPs with variants exhibiting improved brightness and contrast relative to single FP.

Figure: Our Tat_GFP_RFP construct.
Protein G is a transmembrane protein (se figure below) found on the surface of the opportunistic pathogen Streptococcus dysgalactiae ssp. equisimilis) group C and G [10]. The fully sequenced protein G gene comes from S. dysgalactiae ssp. equisimilis) strain 148 and has a gene length of 1679 bp and a protein size of 593 amino acid and a mass of 63.292 kDa [11]. Protein G is usually not a fixed size and can vary from strain to strain [12]. The main reason that Protein G is attractive for our project is its ability to bind to human serum albumin (HSA), the most abundant protein in the blood. This binding has proven to help masking the S. dysgalactiae ssp. equisimilis) cells from the immune system by creating a outer layer of HSA [10]. Protein G is therefore considerd to be involved in, but not the determinator of pathogenesis.

Figure: Protein G binds to human serum albumin (HSA) and masks S. dysgalactiae ssp. equisimilis) from the immune system.
Transport of proteins is an essential part of cellular life. Proteins destined for transmembrane transportation
are usually synthesized with amino-terminal signal sequences, signal peptides, that direct the proteins to
specific membrane transporter complexes. During the transport/export the signal peptide is cleaved from the
protein.
Gram negative bacteria export numerous proteins into the periplasm. The twin-arginine translocation pathway (Tat pathway) is a protein export, or secretion pathway found in plants, bacteria, and archaea. This transport across membranes is via distinct signal peptides with twin-arginine motifs. In contrast to the Sec pathway which transports proteins in an unfolded manner, the Tat pathway serves to actively translocate folded
proteins across a lipid membrane bilayer. In vitro studies (Clark and Theg 1997; Hyndis et al. 1998) confirms that the Tat system is able to transport fully folded proteins in bacteria. Several tat genes
have been identified, and in E. coli the export pathway requires four integral membrane proteins, TatA, TatB,
TatC and TatE. Other componenets cannot be ruled out. Studies show that a twin-arginine signal peptide
is able to direct the export of active green fluorescent protein (GFP) in E. coli and that translocation
almost exclusively occur by the Tat-pathway. As GFP is unable to fold in the periplasm it is assumed that its
translocation must occur in folded conformation [13].
Taking this information into account we discussed the possibility of transporting different proteins into the
periplasm of bacteria, and what this may be useful for. As all gram negative bacteria produce outer membrane
vesicles (OMV's) we looked into the content of these vesicles. Was the sorting of proteins random? Could we
direct certain proteins toward them? And what function would that give? Can we use this to our advantage?
Tat targeting signals exhibit a structure, like Sec signal peptides, comprising of three regions. A polar
'n-region', a hydrophobic 'h-region', and a polar 'c-region'. The twin-arginine motif is conserved and always
located between the 'n-', and 'h-region'(Berks, 1996). The 'c-region' often contain proline residues, a
positive charge and an AA cleavage site. It is believed that the positive charge prevents mis-targeting
of Tat signal peptides to the Sec pathway (Bogsch et al., 1997)[14].
A commonly used signal peptide is trimethylamine N-oxide reductase (TorA) from E. coli. The TorA signal
peptide is 39 aminoacids long giving this base sequence of 117 nucletides:
5' ATGGCCAATAACGATCTCTTTCAGGCATCACGTCGGCGTTTTCTGGCACAACTCGGCGGC
TTAACCGTCGCCGGGATGCTGGGGCCGTCATTGTTAACGCCGCGACGTGCGACTGCGGCG
CAAGCG 3'
Primers were designed using??
{|border="1"
!Forward Primer
!Reverse Primer
!Tm
|-
|5'AGAGAAAGAGGAGAAATACTAGATGGCCAATAACGATCTCTTTCAGGCATCACG 3'(AAC)
|5'GAATTCGCGGCCGCTTCTAGATGGCCAATAACGATCTCTTTCAGGCATCACG 3'(AAC)
|72°C
|}
The first 22 bases of the forward primer and the first 20 bases of the reverse primer are linker sequences. The forward primer was designed to make an overlap with the backbone plasmid and the reverse overlaps with the linker on GFP.
TorA has successfully been fused with the coding region of GFP's in previous studies. The construct was
further cloned into a vector plasmid and introduced in E. coli. The construct, when expressed will then
have the GFP attatched to TorA signal peptide which will direct the protein through the Tat pathway into
the periplasm of the cell. In the periplasm the signal peptide is cleaved of. The GFP is then mainly found
in the cell's periplasm, not in the cytoplasm [13].

Figure: Visualization of GFP in the periplasm of the cell using fluorescence microscopi.
When in the periplasm the GFP may also be included in the vesicles as they budd of. The contents of the vesicles vary, but is believed to be regulated in some way. The proteins that reach the periplasm is transported there by signal peptides for a reason. Knowing this it would be possible to transport other proteins to the periplasm and direct them toward the vesicles. As the vesicles are not a replicative unit like bacteria or viruses it may be a perfect transport vessel for drugs, vaccines or other medical applications. This is the quality that we wish to examine further. Can we introduce surface-proteins on the vesicles that mask them from the immune system? What about a cancer cell recognition type of construct? Program the vesicles to release content only when in contact with harmful cells?
The Pm promoter originate from ''Pseudonomas putida''TOL-plasmid pWW0 ( 15 ). The Pm/XylS expression system is positively regulated by m-toluic acid. The m-toluic acid binds to the XylS protein which is constitutively expressed by the Pm/XylS promoter system. The XylS-m-toluic acid complex binds to the promoter, and activates it.

Figure: Overview of how the Pm/Xyls Promotor system funtions. Production of recombinant protein is dependent on access to the indicer m-toluic acid.
There is a positive correlation between the inducer concentration and the protein production. By increasing the inducer concentration, the expression system will be stimulated and produce more product. This opportunity to control the amount of protein produced, makes it an expression system which is often used, especially here at NTNU. It is also shown to work in different bacterias(Pseudomonas subspecies, Aeromonas hydrophila, Aerobacter aerogenes, Serratia marcescens, Erwinia carotovora and importantly, Escherichia coli (16 ).
The properties of the Pm/XylS promoter system, was an important part in making this in to a BioBrick. It could then be used together with our GFP-RFP construct and regulate how much of the dimer got produced.