 "
"
Team:Penn/AssayOverview
Assay Overview
Measuring Methylation
Two techniques have traditionally been employed to measure DNA methylation: Restriction Based. The first, called Combined Bisulfite Restriction Analysis (COBRA), involves chemically converting unmethylated cytosines into uracils (a process called bisulfite conversion), while leaving methylated cytosines intact. Performing PCR that amplifies the region of interest leaves methylated cytosines intact and unmethylated cytosines converted to thymines (Figure 1). Samples are then digested using an enzyme that will only cut the unconverted (originally methylated) cytosines. The enzyme can no longer recognize unmethylated sites, as they are “TG” instead of “CG”. Designing primers for this process is not always feasible, even with the help of advanced algorithms, and the process needs to be optimized each time a new site is to be analyzed. Furthermore, the workflow takes several days, is expensive, and is not high throughput enough to accomodate screening libraries of candidate DNA-binding-domain-methyltransferase fusion proteins. It has recently fallen out of favor because it is difficult to interpret and does not consider all CpG sites, but only ones which fall within a restriction enzyme’s recognition sequence. (Xiong 1997 and Li 2002). Our methylation assay, MaGellin, is also restriction-based but is much simpler than COBRA because it does not require bisulfite conversion of the DNA. This eliminates most of the problems that made COBRA unwieldy.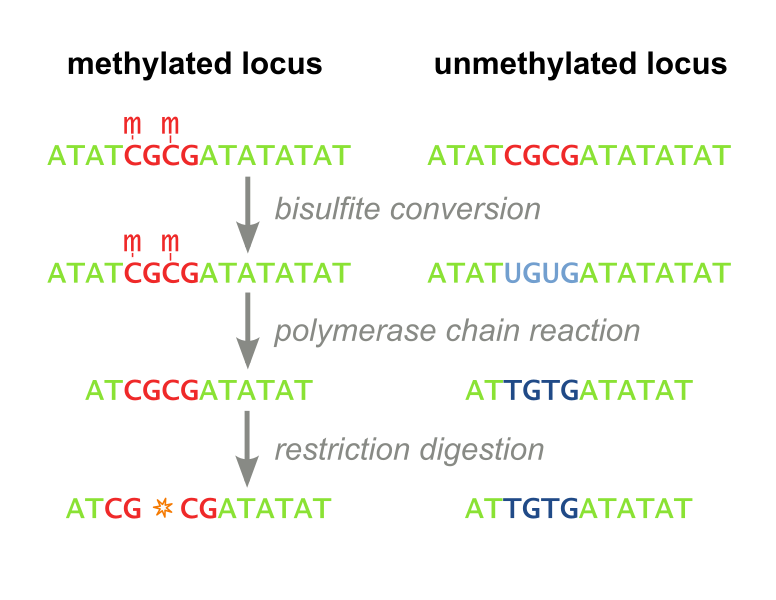

The MaGellin Methylation Assay
Our Team’s Solution.
In order to address the challenges associated with developing new site-specific methylase proteins, our team proposed several different strategies. First, we proposed a migration away from mammalian systems and into E. coli. E. coli does not have a native cytosine methyltransferase, and therefore offers a noise-free environment for methylation studies. Any methylation of CpG sites in E Coli would be a product of a candidate engineered protein rather than the native organism. Second, we envisioned a modular one-plasmid system that can be employed for quickly and cheaply screening the activity and specificity of any DNA binding domain – methyltransferase fusion protein. This plasmid-based methylation assay is called MaGellin.
Plasmid Features.
To accommodate the MaGellin assay, our team designed a plasmid with several key features:- CpG Methyltransferase (M.SssI) with a generic linker sequence in the cloning site. For a working fusion protein and assay, only a DNA binding domain must be cloned into the plasmid. This inherently standardizes MaGellin and lessens the time a user of the assay must spend cloning.
- Multiple cloning site downstream of T7 promoter for orthogonal expression of fusion protein in T7 Express competent E. coli.
- Cloning site for a smaller DNA sequence, specific to the fusion protein being screened – named the “target site”, where the protein will bind. This can be the binding site for a CRISPR-Cas, TALE, Zinc Finger, or transcription factor
- AvaI restriction site 4 bases downstream of the target site – the AvaI restriction enzyme is blocked by methylated CpG sites, thus screening for site specific methylation becomes equivalent to screening for AvaI digestion
- AvaI restriction site sufficiently further downstream of the target site – named the off-target site. This site screens for non-specific DNA methylation as it is spatially removed from where the fusion protein binds to the plasmid.
- XbaI site for linearization of the plasmid. Linearizing the plasmid simplifies analysis of the AvaI digestion by gel electrophoresis.
- Validated bisulfite conversion primer binding sites, so users do not need to go through the time-consuming primer design process if they choose to fortify MaGellin’s results with bisulfite sequencing results
- sgRNA cloning site for users who want to target a CRISPR-Cas binding domain. The sgRNA is constitutively expressed and can be swapped by restriction digest.
- Validated bisulfite conversion primers for users who choose to advance to bisulfite sequencing, for even higher resolution in detecting methylation, after proving their enzyme’s efficacy with our MaGellin assay.
- Kanamycin resistance as a selection marker
Noiseless Chassis.
After cloning this plasmid, we faced the challenge of choosing the correct cell line for the assay. We chose to transform into T7 Express cells for several reasons:- T7 RNA Polymerase in the lac operon allows us to turn on expression of fusion protein after induction with IPTG
- In the T7 Express cell line, genes for several restriction enzymes known to target methylated DNA are knocked out (McrA-, McrBC-, EcoBr-m-, Mrr-). This ensures that our assay plasmid is not cleaved in vivo. Results are difficult, if not impossible, to interpret in the commonly used BL21 cell line.
MaGellin Workflow.
The workflow for screening new fusion proteins with the one plasmid MaGellin bacterial system is as follows:Assemble the MaGellin backbone together with a DNA-binding protein and target sequence of your choosing.
- Forward: CAGGAGGAATTC[ATG] (add start codon only if not included in gene).
- Reverse: CTCTAGAAGCGGC (make sure to remove the stop codon).
- Transform the completed MaGellin plasmid into T7 Express.
- Induce culture with 1 mM IPTG.
- Incubate in a shaker at 37C for 5 hours.
- Miniprep to isolate the plasmid.
- Digest 600 ng of miniprep DNA in a 15 uL reaction with 10 U of both XbaI and AvaI.
- Incubate reaction for 1 hour at 37C.
- Look for 3 distinct band patterns that correspond to specific and interpretable methylation outcomes.
- The presence of large one band corresponds to non-site-specific DNA methylation (AvaI was blocked at both the target and off target sites, and thus only XbaI cut the plasmid)
- The presence of two bands corresponds to site-specific DNA methylation (AvaI was only blocked at the target site, thus AvaI cut in the off target site and XbaI cut the plasmid)
- The presence of three bands corresponds to no DNA methylation – or an inactive fusion protein (AvaI was not blocked at either the target or off target sites and XbaI cut the plasmid)
