 "
"
Team:Shenzhen BGIC ATCG/results
From 2013.igem.org


Playing with my eyes
aren't you?
Hi I am Dr. Mage!
A "budding" yeast cell!
The Magic
Our project “Cell Magic", a complex work has been completed partly,due to the time limitation. The results we have gotten are listed below:
We verified some of the cyclin promoters. The clb6 promoter, which is supposed to be expressed in G1 phase, has been verified perfectly.

Figure. clb6 promoter for G1 phase has been verified
Targeting peptides to three locations have been verified. Others are still being constructed and tested.

Figure. Test for targeting peptides to mitochondria, nucleus and vacuolar.
We got good data for degradation tags for E. coli. Construction of degradation devices for yeast have been finished and being tested. Degradation rate calculated from both methods mates well.

Figure. Pictures for degradation tag testing devices with positive control.
Microfluidics device has been successfully developed for measurement and cell synchronization.

Figure. Cells being captured by microfluidic chip.

Figure. Degradation tags were tested in Microfluidic
Cell synchronization has been successful by changing the method to microfluidics based one.

Figure. All cells are synchronized to G1 phase by using the microfluidics method.
Alternative splicing device are still in construction, related results will be updated later.
Promoter Verification
Achievements
Using the GFP as reporter and morphological alteration as cell cycle representation, we verified the Clb6 can be activated in G1 phase in the yeast.
As shown in the picture in normal lights, there are some yeast were budding. These budded yeast cells and small size budding yeasts (within the red circles) are assumed in the G1 phase of cell cycle. And the next picture was taken under activation light, thus the green lights can representing the Clb6 promoter expressing. The two pictures indicated the Clb6 were expressed in the G1 phase as we expected when designing the experiments
PS: we modified the contrast ratio to lower the lights of neighbor cells, thus our results looking better.
Figure. clb6 promoter for G1 phase has been verified.
We also captured fluorescence on cln3 promoter (S phase) test device, for the fluorescence is quite weak, we utilize micro plate reader and flow cytometry to verify the promoter.

Figure. Flow cytometry to verify S phase promoter cln3.
Problems
Yeast Version
While the promoters are natural ones without optimization, and our shuttle plasmid is pSR416 which is a single copied one, fluorescence is too weak to be captured by microfluidics device. That is a main reason for why we currently cannot show you a perfect cell magic movie. We are now constructing all devices with high copy number vectors, and try to optimize the junction near kozak sequence to make a better magic.
E. coli Version
Promoters for E. coli seems not to be successful yet. In our first version, original 5' utr of promoter's downstream gene is included in BioBrick's sequences. The wild-type promoter and RBS may be too weak, or the junction of RFC[23] may have negative effect to the RBS. So in the present version, original 5' utr has been removed and strong RBS B0030 is used. We will update related results later.
Measurement Devices
Yeast Version







E. coli Version



Reporter Locating
Reporter Modification
Fluorescence proteins have been modified to RFC[23], and stop codons have been removed for fusion protein purpose.

Figure. Fluorescence proteins have been modified to RFC[23], and stop codons have been removed for fusion protein purpose
Targeting Peptides
The four pictures shows the reporters can rightly locate to Mitochondria, Nucleus and Vacuole,respectively. Picture A is the negative control, all yeast cells are lighted with GFP. And figure B is the reporter to the mitochondria, we can saw there are several light spots in one cells. Figure C is the reporter located in nucleus, the green spots are small and there is only one in a yeast cell.The last picture shows the reporter of Vacuolar membrane, the green lights were discrete in cells which was as expected.
Figure. Test for targeting peptides to mitochondria, nucleus and vacuolar.
Degradation Rate
In every curcuits measurement, we firstly test the growth curve through detecting the absorbance. Taken small amount of bateria, inoculate into 400ul LB medium, ensuring the initial concentration between 0.02-0.05 (OD600). Cultured in the room temperature, detect the OD600 value every 20 minutes. Y axis represents the logarithm of bacteria number, X axis represents the growth time.
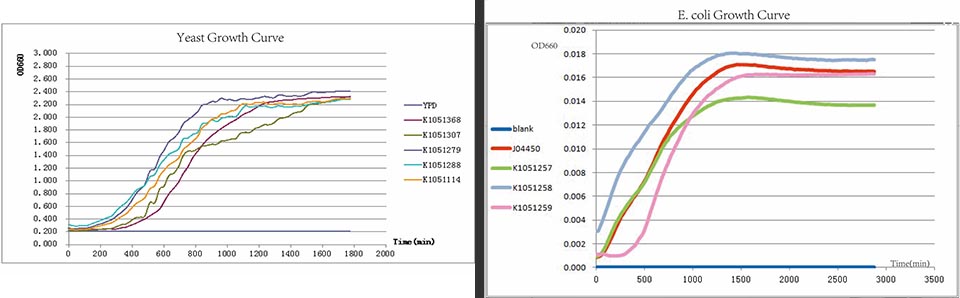
Figure. Growth curves have been drawn for all constructed devices.
In E.coli, the adaptor SspB tethers ssrA tagged substrates to the ClpXP protease, causing a modest increase in their rate of degradation. Which means, a variation of the WT SsrA tag sequence (K1051206, K1051207 and K1051208) will accelerate the degradation of proteins when fused to their C-terminal. Thus the degradation rates are dependent on concentration of proteases and binding mediators.
Achievements
We constructed the measurement pathway of each tag (K1051257, K1051258 and K1051259) to test the rates of degradation of tagged proteins respectively. J04450 was used as positive control because of the same promoter and fluorescent protein.

Figure. Fluorescence ladder for degradation test devices. From left, the negative control,BBa_K1051257, BBa_K1051258, BBa_K1051259, J04450 as Positive Control.
As the pictures showed, the lights of RFP within three degradation tags are decreasing.
Enzyme - labelled meter detect the fluorescent protein intensity
First of all, take a certain amount of bacteria liquid, recovery to around OD0.6(600), ensuring the bacteria in the logarithmic growth phase. Diluted and then the bacteria transferred to 96 well plates, measured their fluorescence intensity. Red fluorescent protein using an excitation wavelength of 584nm, and its emission wavelength is 607nm.When measuring, we first detect the OD600 of each strain, removing the factor of bacteria number difference. Thus the fluorescence intensity cannot be altered by bacteria quantity. Then the measurement mode switching for measurement of fluorescence, fluorescence intensity.
Figure. The test results for K1051257, K1051258, K1051259 and positive control. 1a.Positive control in bright field; 1b.Positive control under exciting light; 2a.K1051257 in bright field; 2b. K1051257 under exciting light; 3a.K1051258 in bright field; 3b. K1051258 under exciting light; 4a. K1051259 in bright field; 4b. K1051259 under exciting light.

Figure. Fluorescence intensity calculated by ImageJ and degradation rate calculated by model of positive control and three tested degradation tags.
The degradation efficiency of three degradation tags are obvious.
While in simulation, we obtained the degradation rate by calculating with [P1_P]/[P2_P]
Figure. The test results of BBa_K1051258 in chip. A,LB medium,O minuts; B, IPTG medium,9minutes; C, IPTG medium, 15 minutes
Using the IPTG medium, the new RFP expression was stopped and we can regard the lights as degradation tags' efficiency. Obviously, the lights are decreasing along with the time. Thus, it indicates that the degradation tags work effectively.

Figure. RFP with degradation tag's half life tested by microfluidics and fit by modeling.
Calculated by ImageJ and from experimental fitting curve we got its half-life of our protein 6.88 min. So the degradation rate should be:

Simulation results calculated and fit from both microscope and microfluidics mates well, which has verified our experimental results.

The datasheet of degradation tags and half-life of E. coli with degradation tags and positive control.
From these data, we can see that the two data from microscope and microfluidic mate very well.
Problems
Yeast Version
The test for yeast degradation tags has not been fully successful. Fluorescence has been captured in some occasions but not stable. It may caused by the same problem with promoter test devices - plasmid copy number.
E. coli Version
Data collected from microscope for different degradation tags did not fully meet performance expectation. Reasons may be multiple to increase systematic errors.
- Fluorescence is too weak for calculating
- Quenching of fluorescence, especially in RFP
- Sample size is small for ImageJ calculating
We will have more data detected by microfluidics and flow cytometry. Results will be updated later.

Cell Synchronization
Microfluidics Device
We made the chip as a platform for watching and synchronize the cells. First step is capture the cells by the chip. As showed in the two figures, both E.coli and budding yeast can be captured by the chip successfully.

Figure. E. coli and yeast cells have been captured by the chip successfully.
As Sic1 method has not been successful, we tried another way by changing medium by microfluidics. Protocols can be found in Notes page.
Figure. All cells are synchronized to G1 phase by using the microfluidics method.
All yeast cells in microfluidics chip are in G1 phase, after budding.
Sic1 Synchronization
Seven phosphorylation sites are mutated by PCR to make Sic1 mutant. However, it has been taking us too long time that we have been still working on right clones. Experiments results will be updated later.
Alternative Splicing
Original Design
In our project, we attempt to use intron as a switch. However, according to previous research, there are two splicing forms of SRC1 intron: one is complete splicing (5’S) while the other can leave 4bp at its 5’ end (5’L). The remaining 4-bp causes frame-shift and a stop codon in the region of adaptor, thus the following exon will not be translated. However, this kind of switch is not complete – the ratio of 5’L and 5’S varied between 40-60 and 85-15.
In order to make a complete switch, we abandoned the remaining 4bp of SRC1 intron to avoid producing 5’L mRNA. So the result comes out to be: when Hub1p are expressed, they bind to spliceosome to modify it, resulting to easy recognizing 5’S splice site and its splicing; when inhibit the expression of HUB1 by CRISPRi system, spliceosome can hardly recognize 5’S splice site, therefore no intron can be spliced.
Further Plans
However, things didn’t go as we expected. Our experiment showed that this 4bp plays important role in SRC1 intron splicing. So, to achieve our goal, we redesigned another three versions of SRC1 intron:
1. Intron with GG at its 5’ end
2. Intron with CGG at its 5’ end
3. Intron with its original 6bp at both end
Due to the lack of time, we are still constructing these parts to test devices.