 "
"
Andrew Crouch
From 2013.igem.org

October 10, 2013
- Did one MAGE cycle with all of our oligos on the strain that was FACS sorted and selected for PLA production
- Attempted to dissolve control PLA in acetonitrile
- No success
September 29, 2013
- Filtered PLA-producing mixture from September 26 and added ~25 volumes of ice-cold methanol
September 26, 2013
- Started cultures of 1, 38, 64, 65, 66 (from frozen stocks) in LB, Nile red, ATC, Arabinose, Kan (3ml LB, 1.5ul NR, 3ul Kan, 3ul ATC, 15ul Arabinose)
- Collected cells that had been producing PLA for about a week in the shaking incubator via centrifugation at 4000 rpm for 20 minutes
- Resuspended them with acetone and left in non-shaking incubator for 20 minutes
- Mixed them with ~20 volumes of chloroform and left in fume hood for 48 hours at room temperature
- Started culture of BL21 for later transformation with Imperial's proteinase K BioBricks
September 15, 2013
- Transformed EcNR2 with the PHA/Pct gene, with the Tokyo BioBrick, and with the pSB1C3 backbone
- Plated on Kan plates
- Transformed BL21 with our BioBrick
- Plated on CAT plates
September 12, 2013
- Started a 1 L culture of EcNR2 with our plasmid with the pLtetO promoter in LB min + kanamycin + arabinose + ATC
- Ran a PCR using the PCT_Bio_F, PCT_Bio_R, bio_PCT_F, and bio_PCT_R primers (numbers 182-185 in frozen stocks) as the forward and reverse primers, and the PCT, Tokyo BioBrick, and pSB1C3 backbone as template
- DPN1 digested the results
- Prepared the Gel Green gel
- Started a BL21 culture
- Started an EcNR2 with our plasmid with pLtetO culture of 3ml LB with 3ul Kan
- Made double stranded DNA mix with 5ul of Spec TetR DNA into 45ul of nuclease-free water
- Went through one cycle of standard MAGE protocol
- Plated on Spec plates and allowed the plates to grow overnight
- DPN1 digested the pLlacO plasmid backbone
- Ran a Gibson assembly with the pLlacO promoter and the backbone
- Started a culture of "wild-type" EcNR2
- Did one MAGE cycle on "wild-type" EcNR2 using the KO oligos, the RBS oligos, and all of the oligos together in three separate reactions
September 9, 2013
- Plate read the 6 cultures started on September 8
- Took 0.5ml from the confluent culture into an eppendorf, spun it for 1.5 minutes, resuspended it in 1ml PBS, spun for 1.5 minutes, and then resuspended in 1ml
- Placed 200ul in each well
- This means there are 5 wells per sample, with another 5 wells with just PBS
- Excited at 530nm and measured at 590nm
- Also, took OD measurements (absorbance at 600nm) of all wells
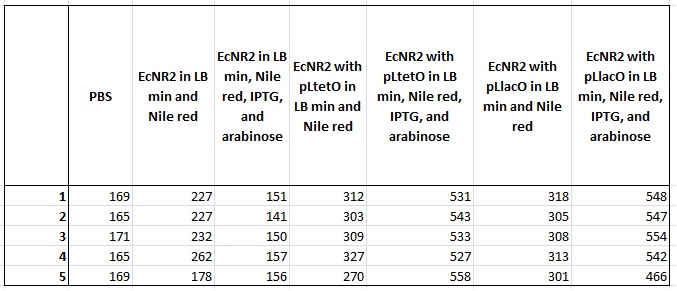
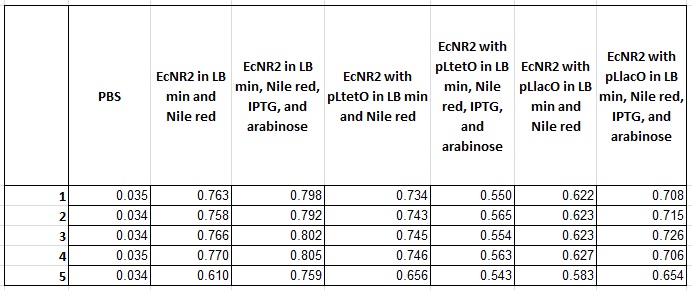
- Made frozen stocks from the other 4 cultures from September 8 (EcNR2 with pLlacO)


September 8, 2013
- Used ice cold methanol to precipitate PLA out of cell debris from chloroform mix made on September 6
- Made frozen stock of Spec TetR 96-well plate
- Each well had a total of 120 uL:
- 20 uL 80% glycerol
- 50 uL LB min
- 50 uL cells
- Each well had a total of 120 uL:
- Started 4 cultures from plates from September 7 entry
- Ran a PCR on each of the 16 cultures started on September 7 by Joel
- Used primers 164 and 165 (in frozen stocks) for a 1.6 kb fragment
- Used 65 degrees C as the annealing temperature and 50 seconds for the extension time, using KAPA HiFi in the mix
- Used a PCT gene as a positive control
- Ran these on a gel
- No bands




September 7, 2013
- Transformed EcNR2 #1 with our new plasmid (where the pLtetO promoter was replaced with a pLlacO promoter)
- Plated on kanamycin plates
September 5, 2013
- Started a 100 mL culture of EcNR2 #1 with our plasmid (frozen stock #38)
- 100 mL LB min
- 100 uL aTC
- 100 uL kanamycin
- 5 mL arabinose
- Ran 4 PCRs using PCT1, PCT4, PCT5, and PCT8 as the respective templates and primers 164 and 165 as the forward and reverse primers, respectively
- Ran on a gel:
- No bands
- Ran on a gel:
- Ran gel with PZE21 without the pLtetO promoter (6.2 kb) and the pLlacO promoter (136 bp)
- Order, from left to right, ignoring the ladders: 4 uL of PZE21 without pLtetO, 20 uL of PZE21 without pLtetO, 4 uL of pLlacO, 20 uL of pLlacO
- Gel purified the fragments:
- Cut the bands out of the gel
- Weigh the gel pieces and assume that mass approximately equals volume
- Add 3x that volume of QG buffer (from Qiagen)
- Place the mixture at 50 degrees C until the gel has dissolved
- Add 1x the original volume of isopropanol and mix thoroughly
- Place in spin column with collection tube and centrifuge at max speed for 1 min
- Discard the flow through
- Add 750 uL PE buffer (from Qiagen) and let sit for 2 min
- Centrifuge, discard flow through, centrifuge, then discard the collection tube and transfer the spin column to an Eppendorf
- Add 30 uL of prewarmed (to 50 degrees C) nuclease-free water and let sit for 1 min
- Centrifuge and discard spin column
- Recombineered the EcNR2 #1 with our plasmid strain (frozen stock #38) with Spec TetR and plated on spec plates
- Also plated positive control (frozen stock #67) and negative control (frozen stock #1)
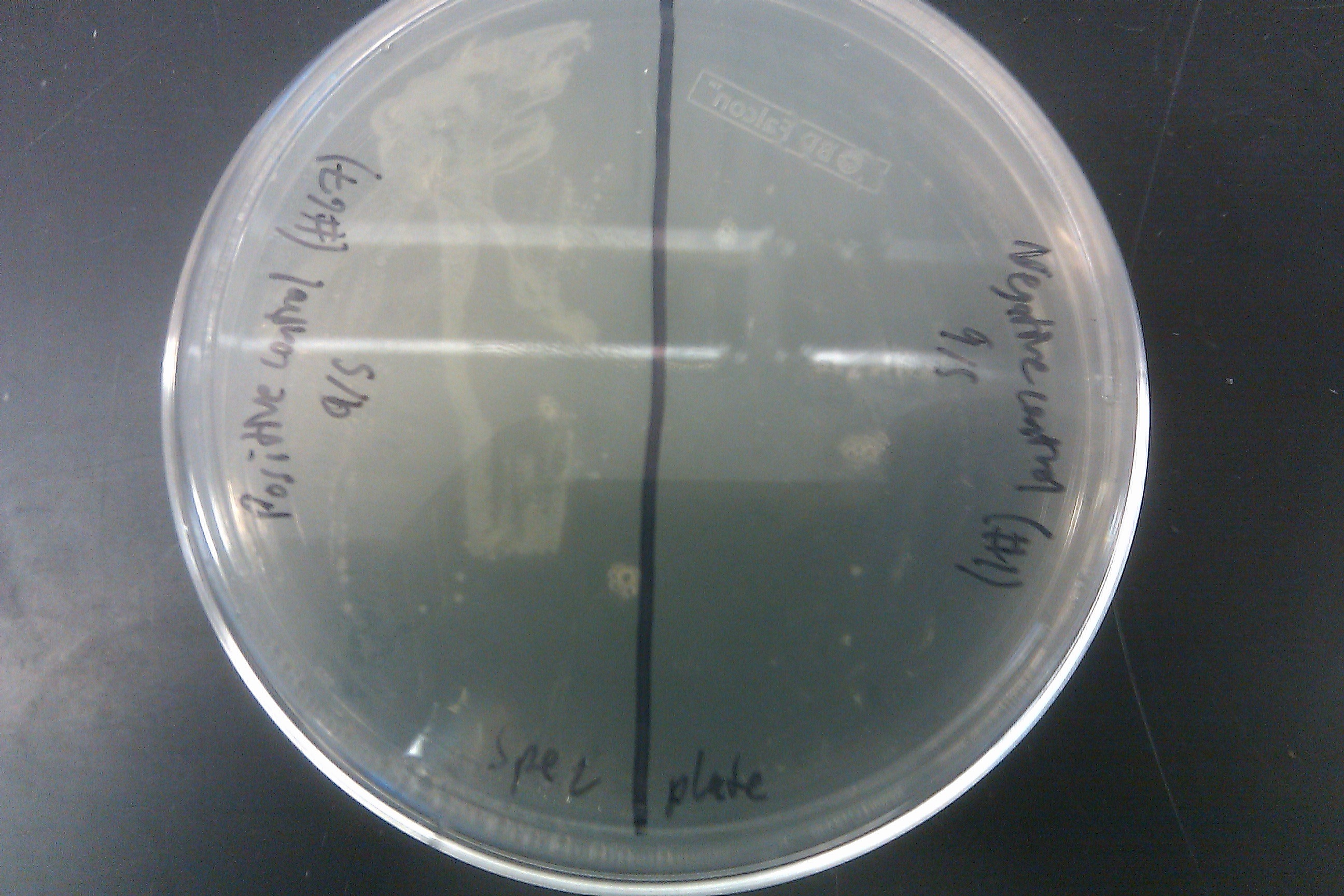
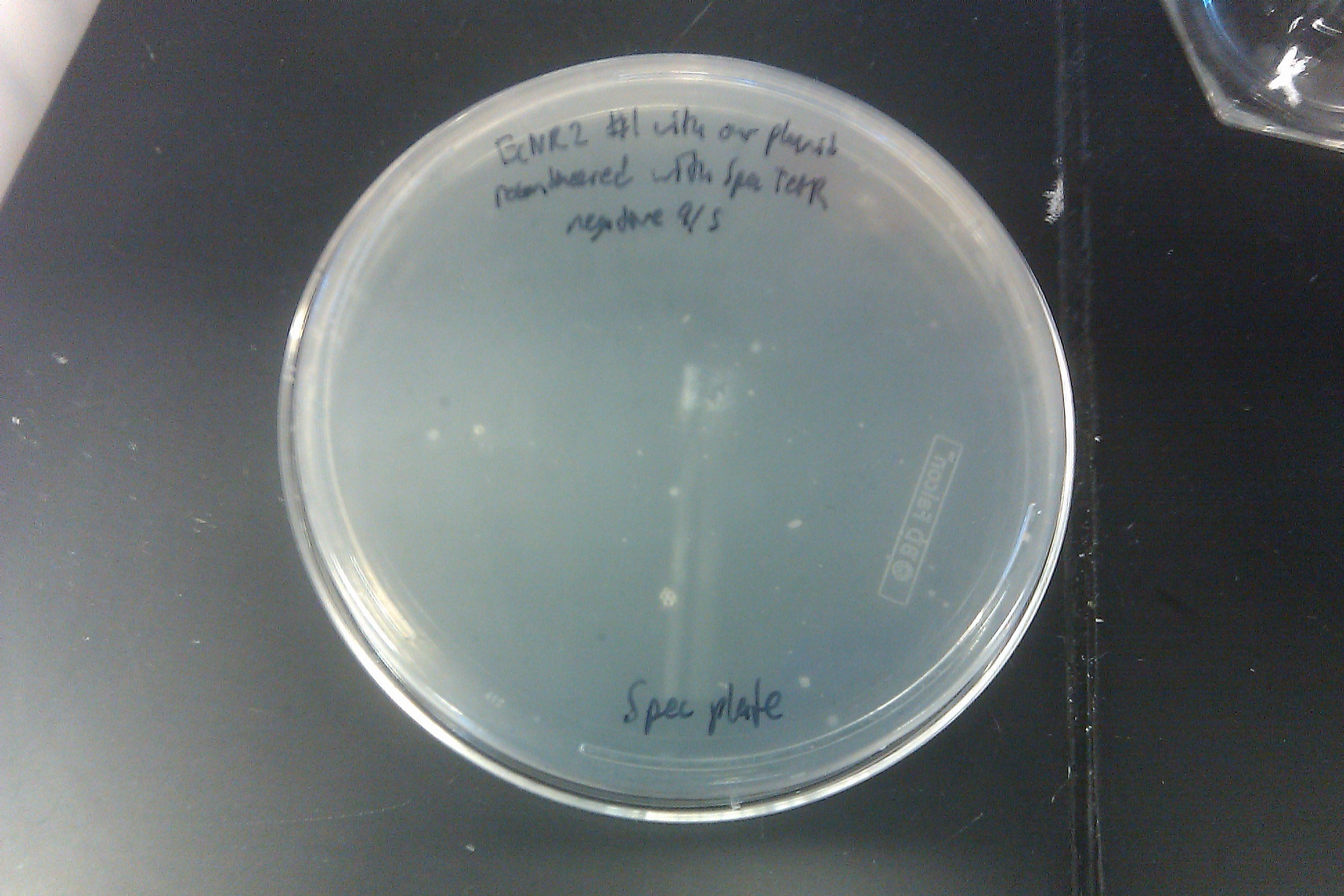
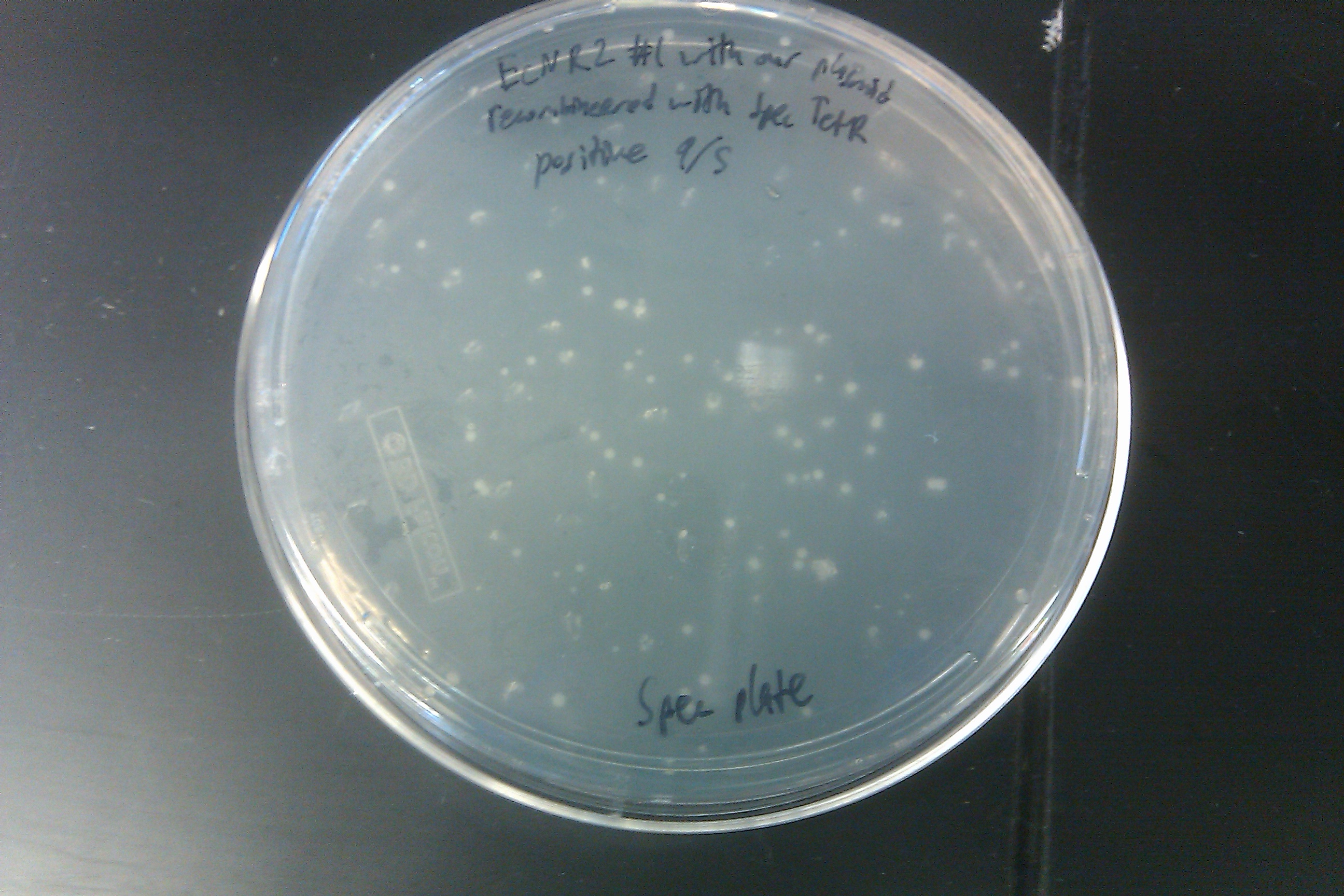
- Also plated positive control (frozen stock #67) and negative control (frozen stock #1)





August 30, 2013
- Retried PCR to amplify Spec with homology to TetR, using primers 172 and 173 and the E. coli Spencer gave us template DNA (see Joel's notebook entry for August 29)
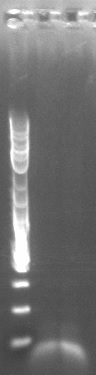
- Still no bands

August 25, 2013
- Ran 1 more MAGE cycle (for a total of 5) on the EcNR2 strain transformed with our plasmid with the KO oligos, the RBS oligos, and all of the oligos listed in the July 31 entry
August 2, 2013
- Ran 1 more MAGE cycle (for a total of 4) on the EcNR2 #1 strain transformed with our plasmid with all of the oligos listed in the July 31 entry
August 1, 2013
- Ran two more MAGE cycles (for a total of 3) on the EcNR2 #1 strain transformed with our plasmid with all of the oligos listed in the July 31 entry
- Did not do separate MAGE cycles for the KO and RBS oligos
- Drop dialyzed the oligo mix
- Noticed that the chloramphenicol media test on the EcNR2 #37 strain transformed with the Tokyo BioBrick from July 31 had actually shown growth overnight

- Since the negative control did not show any growth, it may not be contamination, but rather a sign of an extremely inefficient transformation
- Plated the results on chloramphenicol plates

July 31, 2013
- Ran gel with PCR from July 30 and Joel's experiment from July 30
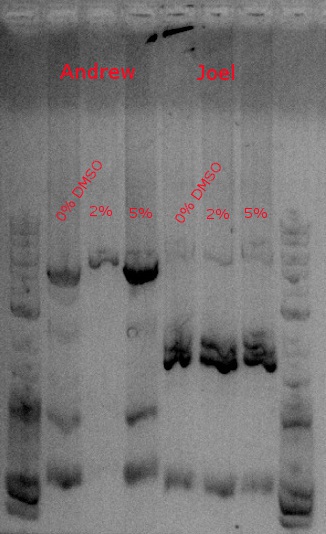
- Results from July 30's transformations:

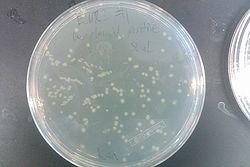
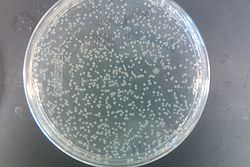
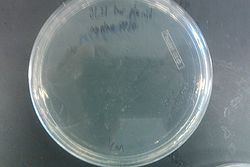

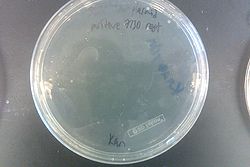

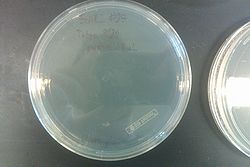

- From left to right, top to bottom: EcNR2 #1 with our plasmid negative control on kanamycin, EcNR2 #1 with our plasmid positive unconcentrated on kanamycin, EcNR2 #1 with our plasmid positive concentrated on kanamycin, BL21 with our plasmid negative control on kanamycin, BL21 with our plasmid positive unconcentrated on kanamycin, BL21 with our plasmid positive concentrated on kanamycin, EcNR2 #37 with the Tokyo BioBrick negative control on chloramphenicol, EcNR2 #37 with the Tokyo BioBrick positive unconcentrated on chloramphenicol, EcNR2 #37 with the Tokyo BioBrick positive concentrated on chloramphenicol
- Started cultures of the EcNR2 #1 with our plasmid positive unconcentrated and the BL21 with our plasmid concentrated
- Nothing grew on the plates with EcNR2 #37 with the Tokyo BioBrick, so did further testing by growing that strain up with media containing chloramphenicol
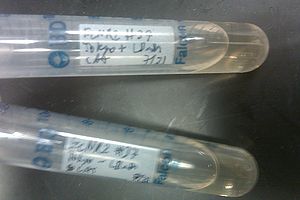
- Negative and positive both show no growth, meaning it was most likely an unsuccessful transformation
- Ran a MAGE cycle on the EcNR2 #1 strain transformed with our plasmid with the following oligos (number in parentheses is the reference number in frozen stocks):
- ackA-KO (1)
- frdB-KO (2)
- frdD-KO (3)
- pflA-KO (4)
- pflB-KO (5)
- adhE-KO (6)
- frdA-KO (8)
- frdC-KO (9)
- ATOB_KO (25)
- PTA_KO (26)
- EUTD_KO (27)
- LDHA_MAG (29)
- ACS_MAG (30)
- ATOB_MAG (31)
- EUTD_MAG (32)
- PTA_MAG (33)
- MAEA_MAG (34)
- MAEB_MAG (35)
- PYKF_MAG (36)
- ENO_MAG (37)
- PGM_MAG (38)
- PGK_MAG (39)
- GAPA_MAG (40)
- TPIA_MAG (41)
- FBAA_MAG (42)
- FBAB_MAG (43)
- PFKA_MAG (44)
- PFKB_MAG (45)
- GLK_MAG (47)
- PGL_MAG (49)
- RPOS_MAG (57)
- Oligo mix was 8 uL of each oligo listed above into 1200 uL total (952 uL nuclease-free water)
- Arced, so tried again using three separate oligo mixes, which were drop dialyzed for 20 minutes beforehand
- One was the one described above ("All")
- One was called "KO" and consisted of:
- ackA-KO (1)
- frdB-KO (2)
- frdD-KO (3)
- pflA-KO (4)
- pflB-KO (5)
- adhE-KO (6)
- frdA-KO (8)
- frdC-KO (9)
- ATOB_KO (25)
- PTA_KO (26)
- EUTD_KO (27)
- This mix was 10 uL of each oligo listed into 1000 uL total (890 uL nuclease-free water)
- One was called "RBS" and consisted of:
- LDHA_MAG (29)
- ACS_MAG (30)
- ATOB_MAG (31)
- EUTD_MAG (32)
- PTA_MAG (33)
- MAEA_MAG (34)
- MAEB_MAG (35)
- PYKF_MAG (36)
- ENO_MAG (37)
- PGM_MAG (38)
- PGK_MAG (39)
- GAPA_MAG (40)
- TPIA_MAG (41)
- FBAA_MAG (42)
- FBAB_MAG (43)
- PFKA_MAG (44)
- PFKB_MAG (45)
- GLK_MAG (47)
- PGL_MAG (49)
- RPOS_MAG (57)
- This mix was 5 uL of each oligo listed into 1000 uL total (900 uL nuclease-free water)
- Made LB min + chloramphenicol plates
- Same as LB min + kanamycin plate protocol, but with 500 uL of chloramphenicol
- Made LB min + ATC + arabinose + Nile red media
- Made TB + ATC + arabinose + Nile red media











July 30, 2013
- Ran a PCR to amplify our construct (PHA and PCT) to give it the biobrick prefix and suffix
- Used primers 148 and 149 and HiFi with 2:30 extension time at 60 degrees (looking for a 4.2kb fragment)
- Running one with no DMSO, another with .5 ul DMSO (2%), and a third with 1.25 ul DMSO (5%)
- 10.5 uL nuclease-free water
- 12.5 uL KAPA HiFi
- 1 uL template DNA (second row, first sample of DsDNA2 PHA/PCT 1ng/ul)
- 0.5 uL forward primer (148)
- 0.5 uL reverse primer (149)
- Transformed EcNR2 (#1) and BL21 with our plasmid, and EcNR2 (#37) with the Tokyo BioBrick
- Followed same protocol from July 2, except plated the strains with our plasmid on kanamycin plates and the strain with the Tokyo BioBrick on chloramphenicol plates
July 29, 2013
- Made LB min media; and LB min + ATC + arabinose + Nile red, LB min + ATC + arabinose + Nile red + glucose, TB + ATC + arabinose + Nile red, and TB + ATC + arabinose + Nile red + glucose plates
- ATC has same concentration as IPTG in July 26 protocol
- Everything else is same concentration as in July 26 protocol
- Ran HiFi PCR on three cultures begun on July 28, Tim's control from July 26, and another colony not picked from Tim's plate from July 25 (done to screen 21B for our construct vs. tolC in post-Kan selection)
- Gathered the necessary materials
- KAPA HiFi
- Nuclease-free water
- Template DNA
- All are EcNR1 mutS Zeo pBAD tolC 21B strains transformed with our plasmid grown in SDS media, plated on kanamycin plates, and then grown in kanamycin media, except for the control, which is not transformed with our plasmid but has kanamycin resistance (?), and the extra colony, which was not grown in kanamycin media (but was still plated on a kanamycin plate)
- Forward and reverse primers
- 21B_F (number 114 in the frozen stocks) and 21B_R (115)
- Mixed the materials together in five PCR tubes, each with 25 uL total
- 10.5 uL nuclease-free water
- 12.5 uL KAPA HiFi
- 1 uL template DNA
- 0.5 uL forward primer (21B_F)
- 0.5 uL reverse primer (21B_R)
- Ran in thermocycler with 58 degrees for annealing temperature and 2 minutes 45 seconds for extension time (30 seconds per kb, expected band of 5.5 kb)
- Prepared gel
- 1.5 g agarose
- 150 mL TBE
- 1.5 uL ethidium bromide
- Ran PCR results on gel at 160 V for about 30 minutes

- Bands at 2 kb are tolC; no bands at 5.5 kb, where our gene construct should have been
- Gathered the necessary materials

July 28, 2013
- Checked the plated recombineered strain from July 26
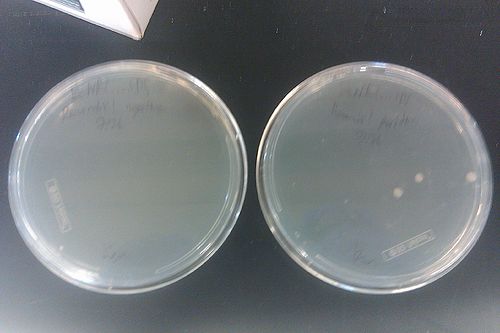
- Left is negative control, right is positive: three colonies grew on the positive, while none grew on the control
- These three colonies were picked and grown in 3 mL LB min + 3 uL kanamycin media
- Left is negative control, right is positive: three colonies grew on the positive, while none grew on the control

July 26, 2013
- Made TB + IPTG + arabinose + Nile red + glucose plates
- 500 mL DI water
- 23.8 g TB powder
- 2 mL glycerol
- 7.5 g agar
- 500 uL 0.1 M IPTG
- 5 mL 20% arabinose
- 50 mL 20% glucose
- 250 uL 1 g/L Nile red
- Grew up EcNR1 mutS-KO Zeo pBAD tolC 21B in SDS, then did one cycle of recombineering with our construct
- Plated on LB min + kanamycin plates
July 25, 2013
- Made LB min media
- Failed at using plate reader 3 times
July 24, 2013
- Made LB min + kanamycin plates
- 10 g tryptone
- 5 g yeast extract
- 5 g NaCl
- 1000 mL DI water
- 15 g agar
- 250 uL kanamycin (final concentration 0.05%)
- Ran miniprep on BBa_K208029 and BBa_K208030 BioBricks cultures grown up July 23
- Finally managed to get several pictures off of my phone:
- Second colicin attempt, this time checking after about 5 hours instead of letting the colicin degrade by growing the cultures overnight

- Left one has tolC, right one does not. As expected, the cells with tolC did not become confluent in the presence of colicin, whereas those without tolC did, confirming tolC's presence in the former
- Tokyo BioBrick transformation (Joel's from 7/19, since Tim's had no fluorescence)
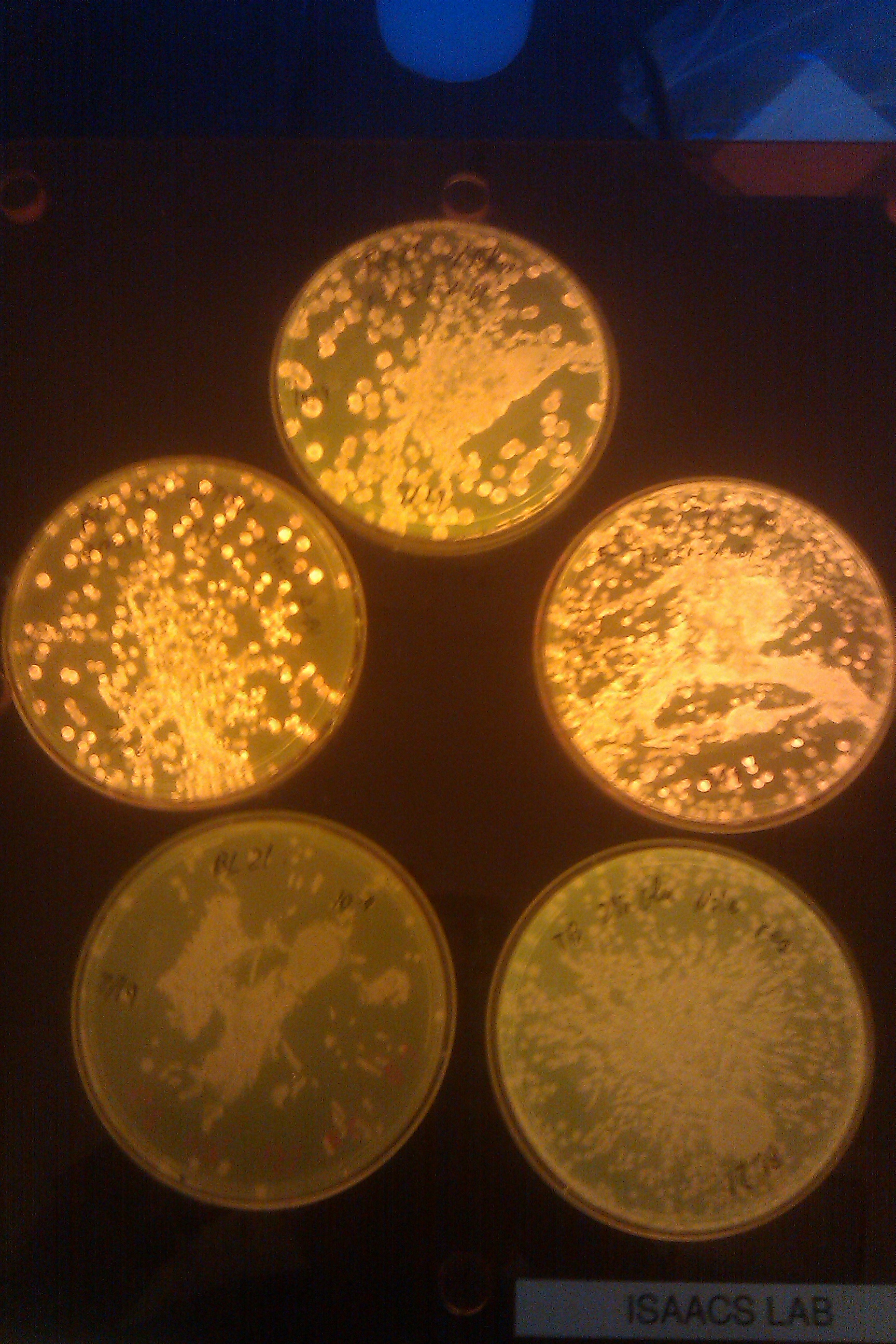
- From top, going clockwise: BL21 with Tokyo, BL21 with Tokyo (big), TB 2% Glucose Nile red, BL21, BL21 with Tokyo grown in TB
- Since only the transformed colonies fluoresced, it is likely the transformation was successful and they are producing P(3HB)
- Second colicin attempt, this time checking after about 5 hours instead of letting the colicin degrade by growing the cultures overnight


July 23, 2013
- Grew up two new cultures of EcNR1 PBAD tolC 21B (number 9 in frozen stock strains) and EcNR1 w/o tolC (10) in 1 mL of LB min and 25 uL of colicin
- Cultures from July 22 were both very confluent, suggesting possible contamination given the success of the SDS experiment done by Joel
- Actually, turns out colicin degrades after 10 to 12 hours, so that would explain it
- Used only 25 uL of colicin in each this time because we only had 50 uL total left
- Cultures from July 22 were both very confluent, suggesting possible contamination given the success of the SDS experiment done by Joel
- Made IPTG + arabinose + Nile red + kanamycin plates
- 500 mL DI water
- 5 g tryptone
- 2.5 g yeast extract
- 2.5 g NaCl
- 7.5 g agar
- 0.0117 g IPTG (final concentration 0.1 mM)
- 1 mL arabinose (final concentration 0.2%)
- 250 uL Nile red (final concentration 0.5 ug/mL)
- 250 uL kanamycin (final concentration 0.05%)
- Made IPTG + arabinose + Nile red medium
- 500 mL DI water
- 5 g tryptone
- 2.5 g yeast extract
- 2.5 g NaCl
- 0.0117 g IPTG (final concentration 0.1 mM)
- 1 mL arabinose (final concentration 0.2%)
- 250 uL Nile red (final concentration 0.5 ug/mL)
- Visualized Joel's and Tim's Tokyo BioBrick (BBa_K934001) plates with dark reader (currently trying to get pictures off phone)
- Joel's fluoresced, Tim's did not
- Picked colonies from each into 3 mL IPTG + arabinose + Nile red medium and into 3 mL LB min
- Also picked Joel's into 3 mL TB
- Grew up new cultures of BBa_K208029 and BBa_K208030 BioBricks (numbers 7 and 6 respectively) each into 3 mL of LB min
- Ran miniprep on Tokyo BioBrick colonies picked into LB min and TB, in addition to EcNR1 pZE21 colonies picked into LB min
July 22, 2013
- Grew up two new cultures of EcNR1 PBAD tolC 21B (number 9 in frozen stock strains) and EcNR1 w/o tolC (10) in 1 mL of LB min and 100 uL of colicin
- Restriction digested the [http://parts.igem.org/Part:BBa_K208029 BBa_K208029] and [http://parts.igem.org/Part:BBa_K208030 BBa_K208030] BioBricks (in plasmid box) with XbaI and SpeI restriction enzymes provided by NEB (NEB protocol found here)
- Gathered the necessary materials
- BBa_K208029 and BBa_K208030
- 10X NEBuffer 2.1
- XbaI and SpeI
- Nuclease-free water
- 5X Orange G
- Made the restriction digest mixes (each total of 50 uL)
- 5 uL 10X NEBuffer 2.1
- 1 uL XbaI
- 1 uL SpeI
- 28/29 uL nuclease-free water
- 15/14 uL BBa_K208029/BBa_K208030
- Realized later that this was 10 times too dilute, but proceeded anyway
- Incubated the mixes for 1 hour at room temperature
- Heat inactivated the mixes at 80 degrees for 20 minutes
- Added 13 uL of Orange G to each mix
- Loaded 25 uL of each mix into a Gel Green gel
- 1.5 g agarose + 150 mL TBE heated until boiling, then allowed to cool, then 15 uL Gel Green added, then poured and allowed to solidify
- Ran gel in TBE buffer at 180 V and then at 166 V for an unspecified length of time
- Voltage was changed without my knowledge
- Gel imaged in Gel-Doc
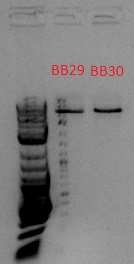
- Bands expected to be at 918 and 1057 bp, but these seem to be at ~5 kb, so it probably wasn't cut
- Gathered the necessary materials

July 19, 2013
- Ran a HiFi sequencing PCR and PCR purification on wells A2, A3, and B4 from July 18
- Gathered the necessary materials
- Template DNA from A2, A3, and B4 cultures
- 1 uL of cells in 20 uL of MilliQ water
- HiFi
- Nuclease-free water
- Primers
- adhE, ackA, pflA, and pflB forward and reverse primers (numbers 29, 30, 31, 32, 33, 34, 35, and 36)
- 12 PCR tubes
- Template DNA from A2, A3, and B4 cultures
- Mixed 12.5 uL HiFi, 10.5 uL nuclease-free water, 1 uL template DNA, 0.5 uL forward primer, and 0.5 uL reverse primer into each PCR tube
- First four received A2 DNA, second four received A3 DNA, and third four received B4 DNA
- First tube in each group of four received adhE forward and reverse primers, second received ackA primers, third received pflA primers, and fourth received pflB primers
- Placed in thermocycler with an annealing temperature of 58 degrees and an extension time of 20 seconds
- This was done because the primer melting temperatures were all just above 60 degrees, and they were all about 500 bp long (~30 seconds per kb)
- Made ethidium bromide gel
- Same protocol as July 4 but with 1.5 uL ethidium bromide into 150 mL of TBE mixed with 1.5 g agarose
- Loaded 4 uL of each tube from thermocycler in gel with 1 uL of Orange G and ran it
- Gel failed
- PCR purified the remainder
- Mixed 105 uL of Buffer PB from the Qiagen PCR purification kit with the 21 uL in each tube
- Transferred the contents of each tube to a spin tube within a collection tube
- Spun the tubes for 1 min at max speed in a centrifuge
- Discarded the flow-through
- Added 750 uL of Buffer PE to each spin tube within a collection tube
- Spun twice for 1 min at max speed, discarding the flow-through after the first time and the entire collection tube after the second
- Placed the spin tubes in separate eppendorfs and added 30 uL of nuclease-free water to each
- Spun those for 1 min at max speed, then discarded the spin tubes, keeping the eppendorfs
- Nanodropped 2 uL from each eppendorf to determine the concentration of the resulting DNA
- Prepared the samples for sequencing
- Added the correct amounts of template DNA, forward or reverse template primer (A for forward, Z for reverse), and nuclease-free water to 24 sequencing tubes
- Submitted the tubes for sequencing to Keck
- Gathered the necessary materials
July 18, 2013
- Ran a MASC PCR on the plated cells from July 17
- Same protocol as July 3, but with only 4 uL of 6x xylene cyanol in each well, and 4 uL of the ladder, with an additional 1 uL of Orange G in two of the wells containing the ladder
- Even though the cells were only MAGEd for ackA, all four primer sets (adhE, ackA, pflA, and pflB) were used (numbers 34, 36, 37, 39, 49, 52, 57, 58, 63, 64, 65, and 66)
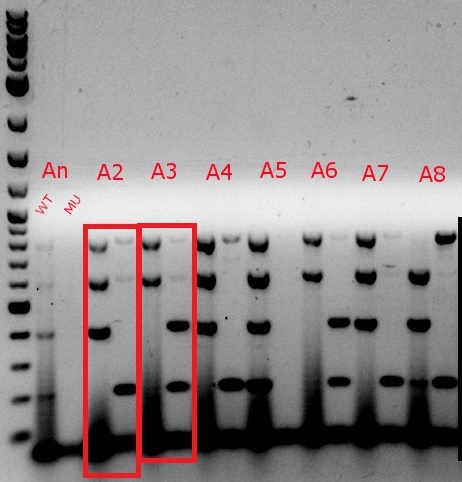
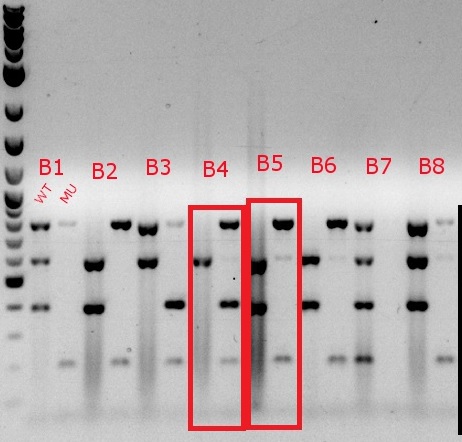
- Boxed wells were grown up in their own cultures overnight


July 17, 2013
- Ran another MAGE cycle on the D8 cells
- This was done because the plates were overgrown to the point where (and the colonies were so small that) it would be extremely difficult to pick just one colony
- Plated using the streaking method described in the July 2 entry on LB min plates immediately after completing the fourth cycle
- Left in 32 degree non-shaking incubator overnight
- Transformed a BL21 strain with iGEM Registry part BBa_K934001 (well 11C in Kit Plate 2), made by the group iGEM12_Tokyo_Tech (specifically, designed by Taku Nakayama), called "phaC1-A-B1 [P(3HB) synthesis]" using the same transformation protocol as on July 2
- Used chloramphenicol plates instead of kanamycin plates
- With bead plating method, plated 1 mL instead of 50 uL
- See full details of phaC1-A-B1 [http://parts.igem.org/wiki/index.php?title=Part:BBa_K934001 here]
- Diluted the iGEM part DNA with 10 uL of nuclease-free water beforehand
July 16, 2013
- Ran 3 MAGE cycles on the D8 cells (which are the result of the 5 previous MAGE cycles; the 6th and 7th were never used) with ackA alone in the oligo "mix"
- Used the same protocol as on July 1 but used nuclease-free water instead of Millipore water in the oligo mix
- Plated using the bead method described in the July 2 entry on an LB min plate soon after completing the third cycle (well before mid-log)
- Left in 32 degree non-shaking incubator overnight
July 15, 2013
- Made IPTG + arabinose + Nile red LB min plates
- 500 mL diH2O
- 5 g tryptone
- 2.5 g yeast extract
- 2.5 g NaCl
- 7.5 g agar
- 100 uL IPTG (final concentration 0.1 mM)
- 4 mL arabinose (final concentration 0.2%)
- 250 uL Nile red (final concentration 0.5 ug/mL)
July 10, 2013
- Ran a gradient PCR with the chosen cultures from July 9 (A9, A11, and D8, as well as the ancestor, D12) with wild-type primers, mutant primers including the newer ackA mutant forward primer used in the July 9 PCR, and mutant primers including the older ackA mutant forward primer used in PCRs before that
- This was done because the ackA mutation was not present in any of the mutant lanes in the July 9 PCR

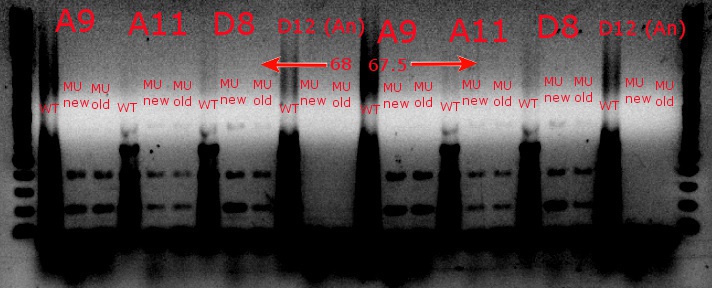
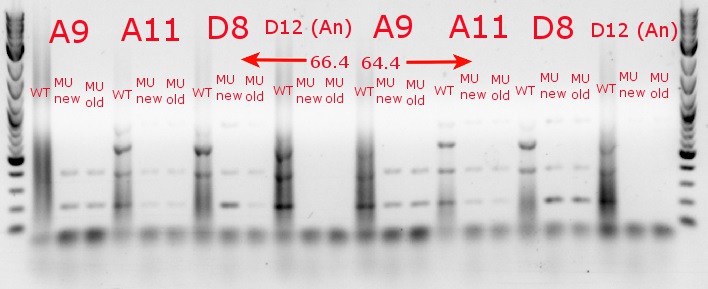
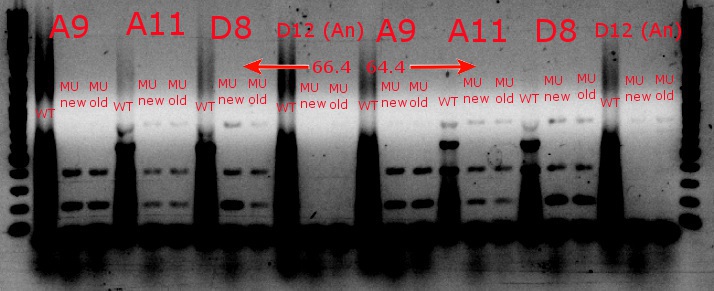


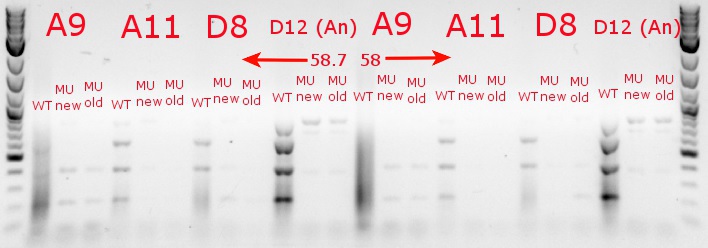
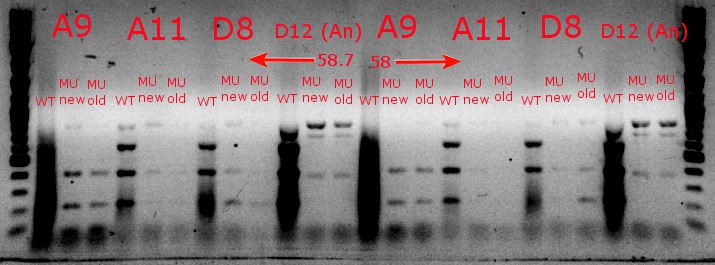
- The images on the left are lighter to see the individual bands inside the streakier lanes; those on the right are darker to see the fainter bands more easily
- This was done because the ackA mutation was not present in any of the mutant lanes in the July 9 PCR








July 9, 2013
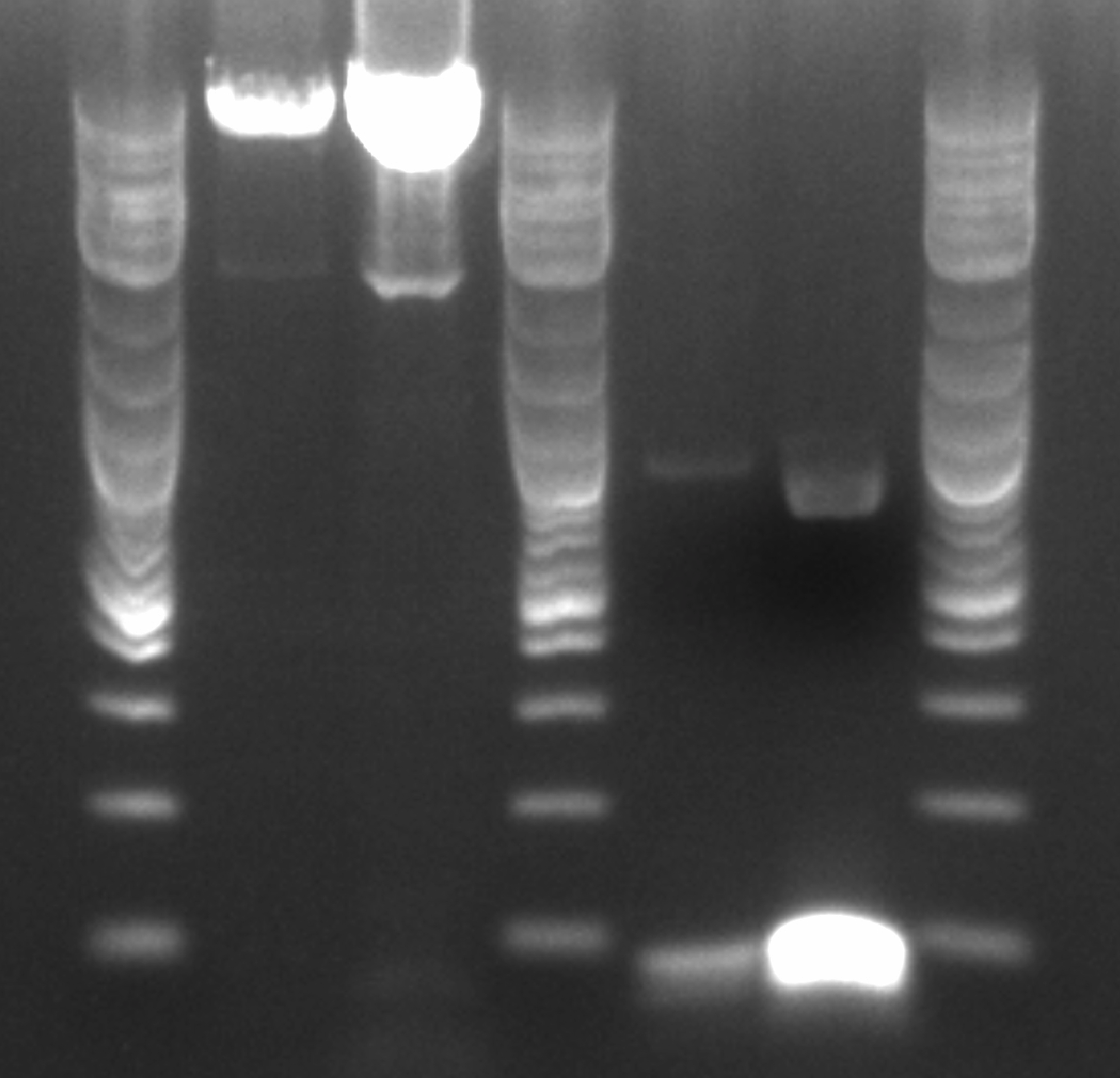
- Ran a gradient PCR using template DNA from the six boxed lanes on the MASC PCR image from July 8: A9, A11, A12, D2, D8, and D12 (the ancestor)
- Used redesigned forward primers for the mutant primers that have Tms closer to their wild-type counterparts
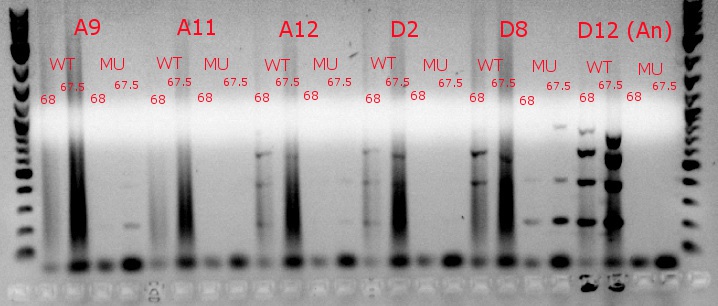
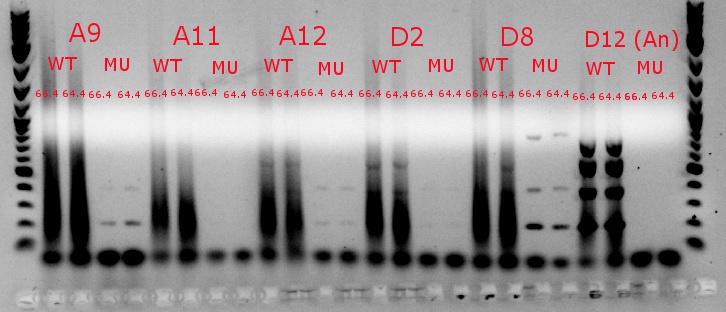
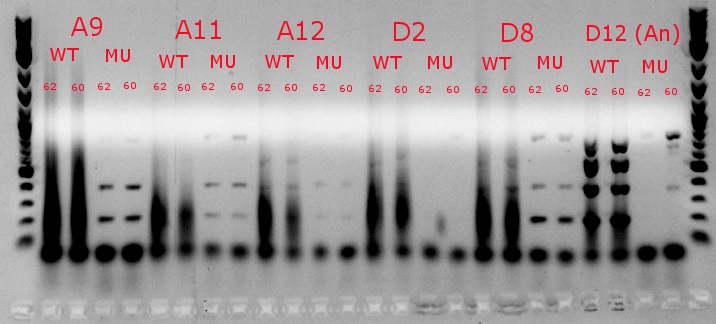
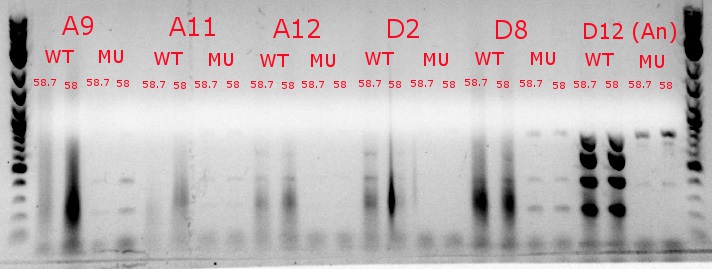
- Analysis here
- Chose A9, A11, and D8 to continue growing
- Used redesigned forward primers for the mutant primers that have Tms closer to their wild-type counterparts




July 8, 2013
- Ran another gradient and MASC PCR, this time with the correct cell and primer combinations (WT cells + WT primers, WT cells + MU primers, MU cells + WT primers, and MU cells + MU primers)



- Top left and top right: numbers denote degrees Celsius
- 60 degrees was chosen as the best temperature
- Bottom: number is the column, letter is the row from the picked-colony cultures; other markings are the mutations observed
- Top left and top right: numbers denote degrees Celsius
July 5, 2013
- Ran another gradient PCR
- See up to step 14 in the MASC PCR protocol on July 3, with the following changes:
- EcNR1 mutS Zeo pBAD tolC 21B was still used as the template DNA for the wild-type PCR tubes, but a strain that had been MAGEd 6 times was used as the template DNA for the mutant PCR tubes this time
- 4 uL instead of 5 uL were used for the DNA ladder
- 1.5 uL of ethidium bromide was added to the agar mix after it cooled but before it was poured into the cast
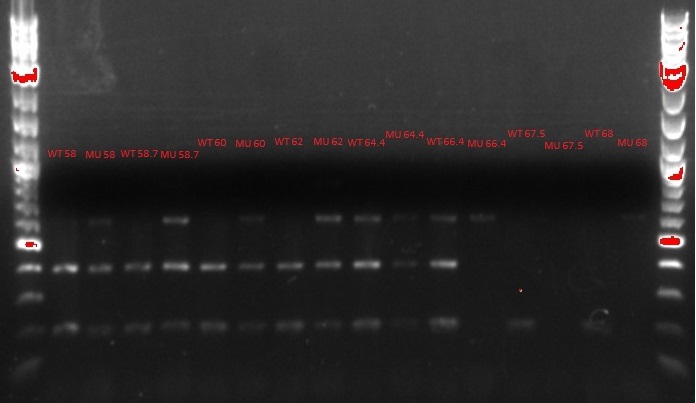
- The xylene cyanol seems to be running at 800 bp, not 2 kb as it should
- See up to step 14 in the MASC PCR protocol on July 3, with the following changes:

July 4, 2013
- Ran gel of PCR plate left in -20 degree overnight, twice
- Mixed 3 g of agarose with 300 mL of TBE
- Microwaved for about 2.5 min
- Let cool
- Mixed in 3 uL of ethidium bromide
- Mixed 5 uL of 6x xylene cyanol into each well of the 96-well PCR plate
- Pippetted 8 uL of each plate well into the corresponding gel well
- Pippetted 5 uL of DNA ladder (already containing xylene cyanol) into the first and last gel well on each comb (total of 4 combs)
- Mixed 1 uL of Orange G into each gel well containing the xylene cyanol DNA ladder
- This was done to better visualize the movement of the 200 bp band
- Put the gel in the gel electrophoresis machine at 180 V for about 40 min
- Imaged gel
- No image available because no good image resulted from this, but it seemed that no band appeared in any of the mutants, which could mean that there is an issue at one of the steps or that we did not do enough MAGE cycles (although the mathematical models support our number of cycles)
- Redesigned the mutant forward primers to have annealing temperatures closer to the wild-type versions of those primers
- Will also decide on a different annealing temmperature, most likely 62
July 3, 2013
- Picked colonies from the streaked positive EcNR1 mutS Zeo pBAD tolC 21B (MAGEd 5 times) plate into 48 wells of a 96-well V-bottom plate, each with 20 uL of MilliQ water, then transferred the contents of those wells into 48 wells of a 96-well flat-bottom plate, each with 150 uL of LBmin with 0.1% Zeocin
- Preparing to perform MASC PCR
- Left plate in shaking incubator at 32 degrees




- Ran another MAGE cycle on the same strain
- Ran a MASC PCR
- Created the primer mix
- Two Eppendorf tubes: MPW1 (MASC PCR wild-type forward + reverse primer 1) and MPM1 (MASC PCR mutant forward + reverse primer 1)
- MPW1 contained 50 uL each of (numbers in parentheses denote vial ID; for laboratory reference only): ackA WT F (34), ackA R (36), adhE WT F (37), adhE R (39), pflA WT F (49), pflA R (57), pflB WT F (52), pflB R (58)
- MPM1 contained 50 uL each of: ackA MU F (35), ackA R (36), adhE MU F (38), adhE R (39), pflA MU F (50), pflA R (57), pflB MU F (53), pflB R (58)
- Two Eppendorf tubes: MPW1 (MASC PCR wild-type forward + reverse primer 1) and MPM1 (MASC PCR mutant forward + reverse primer 1)
- Mixed 5 uL of nuclease-free water and 10 uL of 2x KAPA Fast Multimix into 16 PCR tubes
- Mixed 4 uL of the wild-type primer mix (MPW1) into 8 of them, and 4 uL of the mutant primer mix (MPM1) into the other 8
- Mixed 1 uL of the EcNR1 mutS Zeo pBAD tolC 21B DNA into each PCR tube
- Ran the PCR tubes through a gradient PCR, using the following temperatures during the annealing step: 68, 67.5, 66.4, 64.4, 62, 60, 58.7, 58
- Created the gel
- 1.5 g of agar with 150 uL of TBE, microwaved for 2 min, let cool, then poured into a cast with a 26-well comb
- Placed the gel into the gel electrophoresis machine covered in TBE
- Mixed 5 uL of 6x xylene cyanol into each of the PCR tubes
- Loaded the PCR tubes into the gel, using the outside wells for the DNA ladder (also containing 6x xylene cyanol)
- 8 uL of the PCR tubes in each well, 5 uL of the ladder
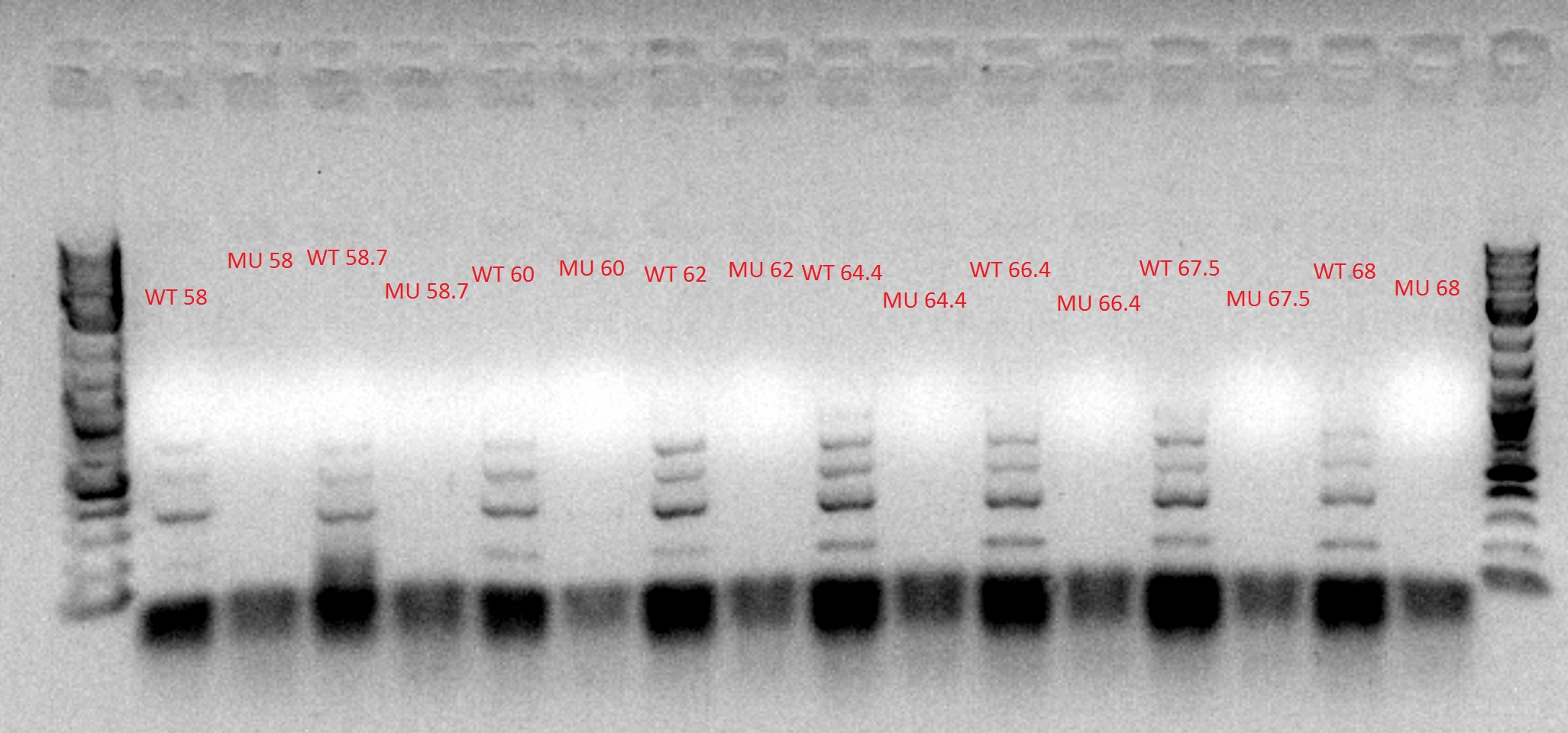
- Chose 64.4 degrees as the optimal temperature, based on the figure above
- 8 uL of the PCR tubes in each well, 5 uL of the ladder
- Made the same mixture as in the PCR tubes in all 96 wells of a 96-well PCR plate, alternating rows such that rows A, C, E, and G were wild-type and rows B, D, F, and H were mutant
- Used the 48 wells of the picked colonies as the template DNA, using each row of the picked colonies plate for two rows of the PCR plate (i.e. row A of the colony plate into rows A and B of the PCR plate, row B of the colony plate into rows C and D of the PCR plate, etc.)
- Ran the PCR plate through a PCR, using 64.4 degrees as the annealing temperature
- Left in the 4 degree step overnight
- Discovered later that it had been moved at the end of the day to the -20 degree freezer overnight
- Left in the 4 degree step overnight
- Created the primer mix

July 2, 2013
- Diluted the results from the MAGE on July 1 by 10x, putting 300 uL of the cells into 3 mL of recovery media
- This was done because the cells were left overnight in the incubator, so they were past mid-log phase
- Made frozen stocks of the dilutions, putting 1 mL of the dilution into glycerol and freezing at -80 degrees
- Frozen stocks #12 for positive and #13 for negative control
- Continued MAGE from July 1 using the dilutions, using the same protocol but leaving the cells in the incubator for about 2 hours in between cycles (4 more cycles completed, for a total of 5 cycles)
- Made frozen stocks after each cycle (14, 16, 18, and 20 for positive, 15, 17, 19, and 21 for negative controls)
- Streaked the EcNR1 mutS Zeo pBAD tolC 21B cells onto a plate with no antibiotic and added 50 uL of each onto another similar plate, using beads to spread the latter
- Left in non-shaking 32 degree incubator overnight
- Transformed Gblock 5 (the minigene) into an XL1-Blue strain (diluted the XL1-Blue by 10x beforehand)
- Prepared minigene by diluting it by 50x with nuclease-free water
- Gathered other supplies
- Nuclease-free water, two Eppendorf tubes, recovery media (1 mL LB min), two electrocuvettes, pipettes and pipette tips, cells
- Spun 1 mL of cells in each Eppendorf at max speed for 1 min
- Removed supernatant, added 1 mL of water, spun again
- Removed supernatant, added 1 mL of water, spun again
- Removed supernatant, added 50 uL of water to negative control and 50 uL of DNA to positive
- Transferred contents of the Eppendorfs to the electrocuvettes
- Wiped off electrocuvettes, pulsed them in the electroporator, then transferred their contents into the recovery media
- Let cells recover 1 hour, then streaked them onto a Kanamycin plate and added 50 uL of each onto another Kanamycin plate, using beads to spread the latter
- Left the plates in the non-shaking 32 degree incubator overnight
July 1, 2013
- Began MAGE on the EcNR1 with mutS knocked out with Zeo (frozen stock #11) to knock out ackA, pflA, pflB, and adhE (oligo numbers 1, 4, 5, and 6)
- Prepared oligo mix
- One uL of each oligo with 46 uL of MilliQ water for a total of 50 uL
- Gathered other supplies
- MilliQ water, two Eppendorf tubes, recovery media (3 mL LB min), two electrocuvettes, pipettes and pipette tips, cells
- Left pipettes and pipette tips in 4 degree cold room
- Heat shocked cells (lids fully closed) for 15 min in 42 degree water bath shaker
- Transferred immediately to ice bucket
- Moved to 4 degree cold room
- Transferred 1 mL of cells into appropriate Eppendorfs
- Spun Eppendorfs in centrifuge at max speed for 1 min
- Removed supernatant, added 1 mL of water, spun again
- Removed supernatant, added 1 mL of water, spun again
- Removed supernatant, added 50 uL of water to negative control and 50 uL of oligo mix to positive
- Transferred contents of the Eppendorfs to the electrocuvettes
- Moved out of the cold room to electroporator
- Wiped off electrocuvettes, pulsed them in the electroporator, then transferred their contents into the recovery media
- Left the cells in the shaking 32 degree incubator overnight (1 cycle completed)
- Prepared oligo mix
June 12, 2013
- Took pictures of the second cycle of MAGE practice
- Plates went bad so all colonies are white
