Overview:
This module is continuation of module 1 and the module mainly consists of specific tumor killer proteins which are expected to decrease cancer cell proliferation and to induce apoptosis. We have two specific apoptotic proteins called Apoptin and E4orf4. In our project plan, engineered bacteria secrete these proteins and these tumor killers penetrate into cancer cells to induce apoptosis within the cells. Through cancer cell penetration, cell penetrating peptides called TAT (Transactivating Transcription peptide) and MPG (designed cell penetrating peptide) have critical roles. Cell penetrating peptides fused with Apoptin and E4orf4 selectively kill tumor cells while normal cells remain undamaged. This point of view takes the advantage of synthetic biology and implies a new targeted therapeutic approach in future.
Goals:=
a.) Apoptin and E4orf4 proteins should be expressed actively and folded correctly.
b.) Apoptotic proteins should pass across the cell membrane
c.) Apoptotic proteins should cause apoptosis and decrease in cell proliferation of cancer cells
Challenges:
1) We want to see the end of cancer cell lives so we chosen apoptosis.Because cells may be eliminated by a number of alternative mechanisms including necrosis. Necrosis is typically described as a “nonspecific” form of cell death, characterized by rupture of the plasma membrane with a consequent localized inflammatory response and damage to surrounding cells and tissues. In contrast, apoptosis is associated with the rapid engulfment and removal of cell corpses by phagocytic cells that recognize “eat-me” signals displayed on the outer surface of the apoptotic cell . Furthermore, apoptotic cell death is the consequence of a series of precisely regulated events that are frequently altered in tumor cells. This provides the opportunity for selective clinical intervention to bring about the death of the tumor cell without damage to normal cells.These basic differences between these two processes of cell death underscore the reason why apoptosis, and not necrosis, represents the most desirable target mechanism for the induction of cell death in tumor cells.
b.)Chosen apoptotic proteins should act as a tumor specific. Despite the success of modern cancer therapies,the three major therapeutic interventions surgery,radiation, and chemotherapy still typically result in considerable damage to healthy tissue.We need new cancer treatments that precisely distinguish between diseased and healthy cells. To this end, synthetic biologists have engineered bacteria to target and invade cancer cells. Engineering bacteria in order to express tumor specific killer proteins enables . We aimed protective healthly cell untill thearapy. Thus we chosen apoptin.
c.) Also we cogitated that proteins alternative to apoptin so we found E4of4. It behave as similar apoptin.In this way we supported more data.
d.) Our constructs can not across membrane because of they are large molecules.So we used TAT peptide.It acts as a membrane shuttle; the structural determinants of transport and the manner by which the peptide crosses the lipid bilayer
e.)We offered that disjunctive penetrating peptide in stead of TAT.So we improve data. Also we selected MPG peptide.
Design and assembly:
We aim to ensure that the specifications that were drawn up are considered in the design.
In recently studies, Apoptin has been described to induce apoptosis in various human cancer cell lines. To develop apoptin as an anti-cancer drug, efficient transduction tools have been used to deliver apoptin into tumor cells. cell-penetrating peptides, such as the trans-acting activator of transcription (TAT) protein transduction domain (PTD) and MPG peptide have therefore been used as vectors for protein delivery.So we decided apoptin fused to TAT peptide because this peptide move apoptin into cell without membrane damage.Also we fused Hemoglutinin A with TAT in order to decrease possibility of getting trapped in microcytosis.In addition to we conceived an alternative technology that offers fusing another cell penetrating peptide with apoptin, thus we constructed MPG peptide with apoptin.And we applied that idea again, on onether protein which names e4of4.E4of4 has been knew induce apoptosis as a tumor specific. We demonstrated e4of4 fused to TAT peptide as a againist tumor cells.As a result, we brought MPG-Apoptin and TAT-E4of4 to literature.
For apply that project, we ordered apoptin fused to TAT, TAT HA and MPG sequences respectively as a composite part in order to minimalize cost and time.And we cloned into pSB1C3 backbone.After cloning, proteins were expressed in BL21 e. coli strain under constutive promoter firstly.İn addition to,our constructs were harboring a 6xHis tag.The 6×His were fused with constructs at the N-terminus because enhance purification.
After cultivatization, we controlled our genes in gel electrophoresis. Electrophoresis showed us the transformation of new genes was successful. There are gel electrophoresis results especially content of Module 2 below:
Experiments and results:
To purify the recombinant proteins, cells were spun down from 50 mL of culture supernatant and resuspended in phosphate buffer saline.PBS contained PMSF in order to prevent proteis from proteases. The mixture was then sonicated on ice eight times for 30 second with a 52% pulsed activity cycle (MISONIX Sonicator W 300). Next, the lysate was centrifuged for 30 min at 10,000 rpm to remove the cell debris. The resulting cell supernatant was loaded onto HIS select affinity column (Sİgma Aldrich)) for protein purification using the standard procedure.. The total protein concentration of each collected fraction from the column was determined using Bradford protein assay with bovine serum albumin acting as the reference protein. The purity of the protein from each fraction was analyzed by 12.5% SDS-PAGE and then the resulting gels were Western blotted using
monoclonal anti-HIS antibody.
Using western blot analysis, it was observed that; our proteins expressed and purified effectively.
Firstly, We offered that using Immunofluorescence staining method for understanding TAT fusing proteins lokalization.Therefore,we fused to TAT-RFP construct with 6xHis tag in order to prove TAT activity.Immunofluorescence is a technique used for light microscopy with a fluorescence microscope and is used primarily on microbiological samples. This technique uses the specificity of antibodies to their antigen to target fluorescent dyes to specific biomolecule targets within a cell, and therefore allows visualisation of the distribution of the target molecule through the sample.When Flourochrome labeled anti Histag antibody bond 6His tag which in front of our construct we visualized TAT-RFP intracellular area.Also we stained DNA and nuclei using Hoechst dye (Thermo Scientific Pierce Hoechst 33342 Fluorescent Stain ) thus we were sure that, TAT peptide alllows penetration .
Investigate whether his-TAT-Apoptin, TAT-HA-Apoptin, TAT-E4of4 and MPG-Apoptin protein expressed by E. coli has apoptotic activity when intro-duced into tumor cells, purified protein was used to examine the protein's apoptotic activity when it was used to treat human colon cancer HT29, breast cancer MCF7 and head-neck cancer cell lines.while we used different factors such as dosage, cell line, changing purified methods we repeated experiments.so we analysed cell viability index under changing factors.We used xCelligence for analysis. The xCELLigence System, also known as RT-CES system, which contains a series of Real Time Cell Analyzer (RTCA), is a labeling free cell based assay system integrating microelectronics and cell biology, suitable for uninterrupted monitoring of biolgical processes of living cells.
We conducted experiments on in vitro cultured HT29 cells which were treated with TAT-Apoptin, TAT-E4of4, TAT-HA-Apoptin and MPG-Apoptin. The results of the X-Cellingence revealed that our constructs was able to significantly inhibit the growth of the HT29 cells. We used only DMEM and elution buffer containing well as a control respectively.
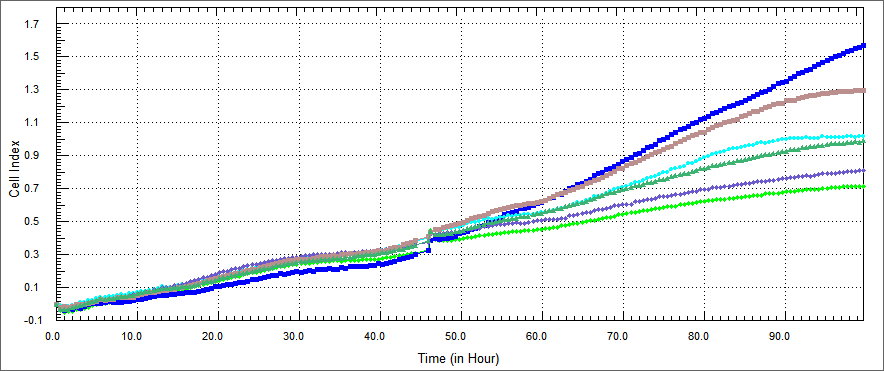 In growth rate of HT 29 cells was performed by xCELLigence after %5 concentration proteins of the purified E. coli-expressed recombinant proteins was co-cultured with HT-29 cell for 54 hours. Non-apoptosis controls, only DMEM and only elution buffer treated cells were assayed. These represent respectively HL-60 cells only (blue), HT-29 cells co-cultured with protein elution buffer (brown) and HT-29 cells co-cultured with TAT-Apoptin (light green),TAT-HA-Apoptin (dark green), MPG-Apoptin (light blue) and TAT-E4of4(purple).
In growth rate of HT 29 cells was performed by xCELLigence after %5 concentration proteins of the purified E. coli-expressed recombinant proteins was co-cultured with HT-29 cell for 54 hours. Non-apoptosis controls, only DMEM and only elution buffer treated cells were assayed. These represent respectively HL-60 cells only (blue), HT-29 cells co-cultured with protein elution buffer (brown) and HT-29 cells co-cultured with TAT-Apoptin (light green),TAT-HA-Apoptin (dark green), MPG-Apoptin (light blue) and TAT-E4of4(purple).
This results observed that in a certain range of concentration and time, our proteins inhibited the proliferation of the HT 29 cells time dependently. Its inhibitory effect was %5 concentration and 54 hours.
Using a similar approach, we tested the apoptosis-inducing ability of our constructs in HEK 293 human kidney cell lines.HEK 293 were co-cultured with TAT-Apoptin, TAT-E4of4, TAT-HA-Apoptin and MPG-Apoptin.In a desired concetration; we expect that; growth curve of treated with our construct HEK 293 cells should be similary growth curve of not treated HEK 293.
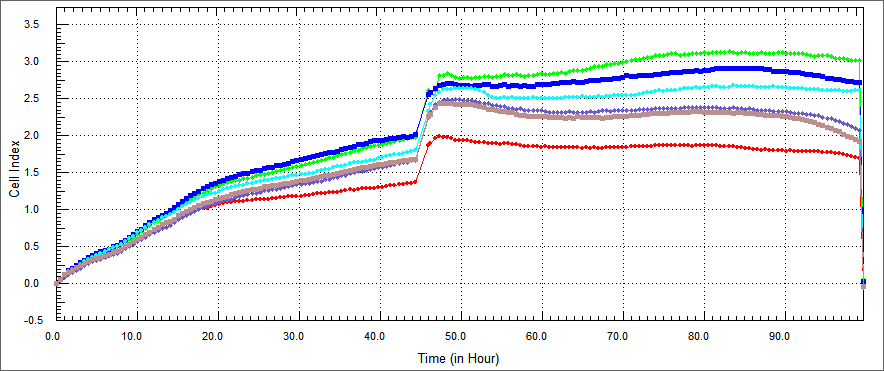
Figure 1:
In growth rate of HEK293 cells was performed by XCelligence after %5 concentration proteins of the purified E. coli-expressed recombinant proteins was co-cultured with HEK-293 cell for 54 hours. These represent respectively HEK-293 cells only (dark green )), HT-29 cells co-cultured with protein buffer ( blue) and HT-29 cells co-cultured with TAT-Apoptin (red),TAT-HA-Apoptin(brown), MPG-Apoptin (light blue) and TAT-E4of4(purple)
Finally we have quantitative data. For supporting this results we need qualitative results.When cells going to apoptosis, they change morphology We made use of this data, we decided take a photos of exposured cancer cells. We seeded colon and breast cancer cells on 12 well plate. Cancer cells co-cultured our proteins at desired dosage.after 24 hours, we took photos.
MCF-7 Breast Cancer Cells Morphology Changes
MCF7 cells morphology changes can be visualized below. Photos are taken 24 h after treating with apoptotic proteins and with 20x focus.

HT-29 Colon Cancer Cells Morphology Changes
HT29 cells morphology changes can be visualized below. Photos are taken 24 h after treating with apoptotic proteins and with 20x focus
Also we thougth other ways of demonstrating apoptosis, we did XTT assay. XTT assay is a colorimetric assay for the nonradioactive quantification of cellular proliferation, viability, and cytotoxicity.We measured spectrophometric analysis by ELIZA. the improved XTT method was used to study the effect of our constructs on the growth of colon, and head-neck cancer cells. The cell concentration was adjusted to 5x104 cells/mland the cells were added to a 96-well plate, at 90 µl/well. There was %10 concentrations of each constructs and each protein was set in three parallel wells with 10 µl/well. The cells were then cultured at 37˚C in a 5% (volume fraction) CO 2incubator and incubated for 48 h. XTT (5 mg/ml) was then added at 50 µl/well and the cells were cultured for 4 h. Following a period of 24, 48 or 72 h, cell viability was determined by a microplate reader at 5 nm single wavelength optical density (OD)
Future aspects:
To carry on the work on the apoptosis module, there are a number of steps we would take in the immediate future and others that form part of our long term plan.
a.)short term plans:
we will perform apoptotic effect of e4of4 and apoptin using alternative methods such as caspase 3 and annexin V.Because of these methods supporting certain data of apoptosis we will establish experimental results.
It is important to, our suc
 "
"


