 "
"
Team:NTNU-Trondheim/ExamplePage
From 2013.igem.org




Traditionally, most sensing systems in synthetic constructs have been borrowed from nature. However, very often researchers want to detect a ligand that does not have a receptor in the existing list of characterized sensors. Scanning natural systems for proteins that bind to a specific molecules is difficult and time-consuming, when it is not impossible. As synthetic biology realizes more and more unnatural functions, we will want to be able to design cells that can detect molecules that life has never before responded to. This is something we cannot achieve using natural receptors.

Here at Berkeley we were motivated to address this problem by constructing a biosensing mechanism that could be readily adapted to the ligand of choice. We wanted to build a modular biosensing device that could be modified by swapping binding domains. This approach would provide a standard method for engineering biosensors that could make use of both natural binding domains, optimized by evolution, as well as engineered domains that respond to molecules outside the range of natural receptors.

ToxR is a characterized transmembrane transcription factor from Vibrio cholerae with active domains in both the periplasm and cytoplasm. Natively, it is involved in the transcription of a number of virulence factors, including the two subunits of the cholera toxin ctxAB and the TCP pilus. It is activated in trans by ToxS, a membrane-anchored periplasmic protein coded by the gene directly downstream of ToxR. Active ToxS in the periplasm stabilizes the dimerization of the periplasmic domain of ToxR. The association passes through the membrane, bringing the cytoplasmic domains of ToxR together to form an active homodimer. The active ToxR dimers drive transcription from the ctx promoter by binding to TTTGAT repeats and recruiting transcription machinery.
ToxR elegantly allows Vibrio cholerae rapid sensing and the activation of pathogenic functions. Virulence factors such as those controlled by this system are often clustered in the genome within pathogenicity islands, remnants from a past horizontal gene transfer. The simplicity and orthogonality of the systems contained in these small virulence cassettes make them an ideal source for modular biosynthetic tools. Indeed, one of the reasons ToxR is so interesting is that it single handedly achieves the task of the standard two-component signal transduction pathway: it is activated in the periplasm and directly promotes transcription in the cytoplasm. The simplicity of this regulation system and the scope of this single protein make it an ideal starting point for synthetic design.

Dimerization of ToxR has been found to activate the ctx promoter in Vibrio cholerae by binding directly to the DNA. DiRita et. al. (1991). ToxS detects changes in the environment, interacts with, and dimerizes ToxR's periplasmic domain. Dimerization of the periplasmic domain of ToxR results in dimerization of the cytoplasmic domain, which activates transcription of the ctx promoter. The ToxR domains are all on one peptide, so signals can be quickly relayed from the periplasm to the cytoplasm. This unique aspect of ToxR makes it a good part for building a biosensor. We aim to make a biosensor by further engineering this dimerization-dependent transcriptional activation feature of the ToxR system to be ligand dependent.



The first step was to validate that a ToxR based two-hybrid system could be expressed in E. coli and that we could cause dimer-dependent transcriptional activation using a chimeric ToxR with periplasmic domains of our choosing. We started by truncating ToxR to eliminate the periplasmic domain because the dimerization state of ToxR's cytoplasmic domain controls transcription. Next, we built a translational fusion of the constitutively dimerizing leucine zipper protein IILK to the ToxR truncation. The constitutively dimerizing protein IILK portion of the translational fusion is located in the periplasm, while the ToxR cytoplasmic domain remains in the cytoplasm. The constitutively dimerizing IILK periplasmic domain should dimerize, causing the cytoplasmic domain of ToxR to dimerize, activating transcription from the ctx promoter.


In order to resolve the toxicity of ToxR, we constructed a new design principle. Stress is a natural regulatory cue in bacteria that dictates changes in behavior or expression levels. We decided to look into the E. coli genome for stress-based regulatory elements. We wanted to find a promoter that would drive transcription while the cell was happy and health, but would shut down in response to cellular stress, behaving as a self-regulating expression system. Toxic over expression of ToxR would be down regulated with this negative feedback system. With this method, we are able to express ToxR chimeras at optimal levels without toxicity to the cell.


Stress is a common regulatory cue that allows organisms to adjust to variable conditions in the environment. As a result, stress-responsive regulatory elements are prevalent motifs in nature. Due to the toxicity caused by the toxR chimeras in E. coli, a method of regulating its expression to produce the maximum amount of ToxR chimera without killing the cell was needed. We hypothesized that the E. coli genome likely contains such stress responsive regulatory systems from which regulatory feedback systems can be constructed. Expression of the ToxR chimera to toxic levels by stress promoters can be downregulated by stress in a feedback loop. This maintains a high but nonlethal level of expression of the toxic gene.

In order to construct a feedback system, we analyzed microarray data of the E. coli genome for regulatory elements downregulated by stress. Moen, et. al. (2009) subjected E. coli to generalized stresses and measured the global mRNA levels of all genome elements for stress response. From their data, we identified 35 potential stress promoters that were downregulated under all stress conditions tested for further characterization. We isolated the region of the genome upstream of the ORF through PCR and placed the resulting pool of 35 stress promoters in front of GFP.

The construct was transformed into E. coli and subjected to general stress conditions. We measured the fluorescence with a tecan plate reader and looked for decreased fluorescence. While not all promoters responded by downregulating GFP expression, a few did show a significant change in fluorescence, with the cold condition yielding the best data. The fluorescence measurements from the Tecan plate reader were confirmed by flow cytometry.


Since some of the stress promoters displayed the desired behavior, we proceeded to incorporate the stress promoter pool into a stress response system with ToxR. In our stress feedback system, expression of the ToxR chimera activates the ctx promoter (Pctx), driving GFP production, and creates cellular stress. The stress promoter responds to stress by downregulating ToxR-IILK expression to a nonlethal level, allowing the cell to maintain a high level of ToxR chimera expression without toxicity.

In order to ensure nonlethal expression of the ToxR chimera, we created an rbs library for each of the 35 stress promoters to construct a promoter-rbs library in front of ToxR-IILK. We transformed the plasmid into cells containing Pctx.RBS.GFP and grew them up to assay the efficacy of the stress responsive feedback system. In our growth assay, we looked for large, healthy colonies as well as strong fluorescence, indicators of robust growth and desired expression levels.

From our combinatorial approach towards the construction of the stress-responsive feedback system, we were able to obtain big, healthy green colonies growing on our plates. The observation of big colonies indicated that our cells were healthy and not experiencing any apparent cellular stress. The high levels of GFP observed in the colonies indicated that cells were expressing enough ToxR chimera to strongly drive transcription of the ctx promoter (Pctx). Since we saw colonies that were big and green, we claim to have successfully resolved the toxicity of ToxR fusion proteins enabling strong transcriptional output while minimizing cellular stress.

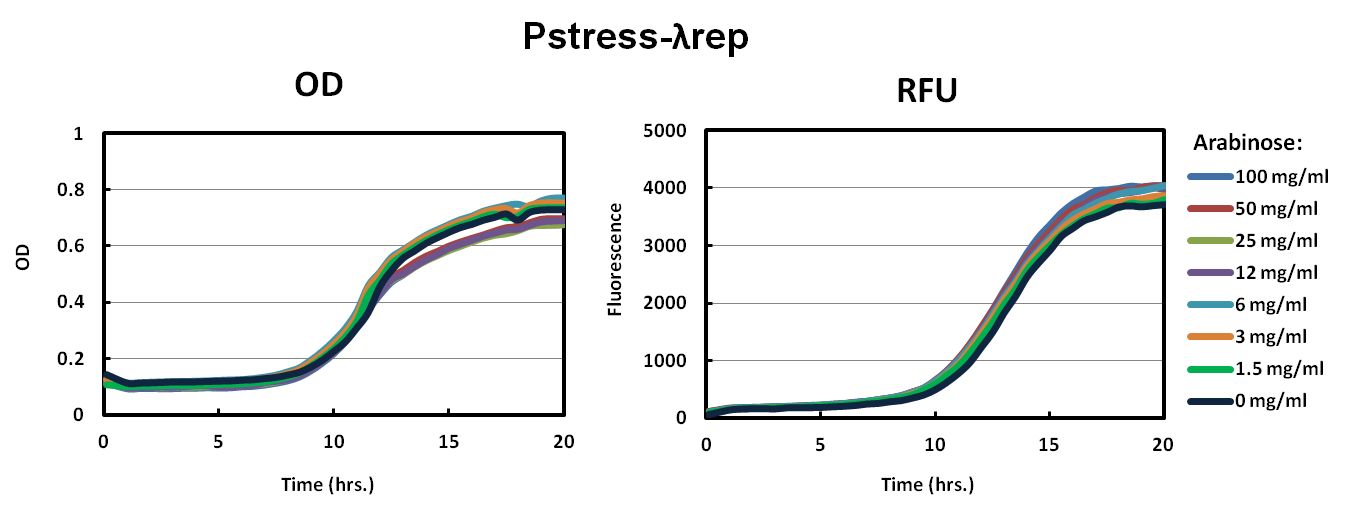
We picked big green colonies and found that cultures showed improved fluorescence and OD with respect to pBAD promoters. In other experiments, we found that Lambda-Rep provided a better fluorescent signal so we replaced the IILK domain with Lambda-Rep in both the pBAD and Pstress, grew them in the same arabinose conditions and measured OD600 and RFU with a Tecan plate reader every five minutes. The as with IILK, ToxR-Lambda-Rep chimeras show improved fluorescence, reduced toxcity, and cells harboring this plasmid behave much more predictably.
From our results thus far, we were able to:
-Characterize a library of stress promoters from the E. coli genome, providing data that they function as stress-responsive downregulators-Create an off-the-shelf negative feedback expression system using our stress promoters that enables the expression of toxic proteins to their highest non-toxic levels, solving our problem of ToxR chimera toxicity
-Demonstrated our proof of principle biosensing system by creating constitutively dimerizing ToxR chimeras that drive transcription off of the ctx promoter (Pctx)
Having achieved our goal of testing our proof of principle, we were ready to construct inducible homodimerizing systems.

Once we had successfully expressed a ToxR chimera that demonstrated constitutive transcription off Pctx we were ready to design a system that could be inducible, and thus a more useful biosensor. We set out to find a ligand dependent homodimer, and after an extensive literature search, we decided to use the Estrogen Receptor. We chose the Estrogen receptor because it fulfilled the criteria of being a homodimer and dependant on Estradiol for dimerization. Furthermore, it has been previously used in a two-hybrid assay as a chimera attached to GAL4 parts to measure ß-galactosidase activity (Peters 1999). In addition to its structural and mechanical advantages it would also make a useful biosensor since estradiol poses numerous hazards to the environment. Therefore the ability to sense it cheaply and effectively would have great implications.
The Estrogen Receptor is made up of 6 domains: A, B, C, D, E, and F. Domains A and B serve as a transcriptional factor, domain C recognizes and binds to specific DNA sequences and is also involved in dimerization (Peters 1999). Domain E is the Ligand Binding Domain (LBD); when it binds to estrogen it homodimerizes to another Estrogen Receptor LBD. Domain D’s function is unknown. Domain F has been shown to inhibit dimerization and/or binding of estrogen and is thus undesirable for our project. In a wild type system these domains interact to bind Estrogen with Domain E, confer conformation so that C can bind a specific DNA sequence, dimerize with another estrogen receptor and act as a transcription factor to promote transcription downstream of its binding.

After studying the function of each of the domains as well as the findings of Peters 1999 paper we began by making two different ToxR chimeras with two Estrogen Receptor truncations: ER∆F (no F domain) and the ER-LBD alone. Both of these estrogen receptor truncations were attached to the 3' end of ToxR and tested under various Estradiol levels. However, these truncations were non-responsive with and without estradiol. We hypothesized that different estrogen receptor truncations may be unable to express in the membrane, and the LBD by itself does not homodimerize as it didn't in the Peters' 1999 study. So, in order to mitigate both possibilities we made 8 more truncations that spanned the range between Domain A and Domain C.

Preliminary data suggests that truncations 5, 6, and 8 all showed constitutive ToxR dimerization and resulting fluorescence. However, none seemed to be inducible in response to Estrogen. Nevertheless, we believe we are getting very close to successfully expressing an inducible chimera because by showing activity it proves the estrogen receptor is not broken. Peters 1999 showed that the estrogen receptor can show dimer inducibility as a chimera in the presence of estrogen, and we think that trying a few more strategies will allow our ToxR chimera system to be inducible. We are going to try several more truncations, as well as various linkers, and the co-expression of different estrogen receptor truncations as was shown to work in the Peters paper.
One issue that could be affecting the Estrogen Receptor is the physiology of the periplasm that causes its cysteines in the LBD to cross react. This could potentially throw off the structure and therefore the functionality of the ER. Ee sought to correct this problem by growing the cells in a reducing environment that would prevent such bonds from forming. Our preliminary results below show that Trunc 8 is greatly stabalized by the addition of beta-mercaptoethanol (BME), effectively creating a reducing environment sensor.

Since the above system, although clearly stabalized in the periplasm, was not estradiol inducible we decided to look deeper into its ability to sense reducing environments. The first experiment showed how it grew in a reducing agent, while our next experiment below showed how it would respond (after growth saturation) to introduction into a buffered and non-buffered reducing environment. Once again Trunc 8 showed impressive inducibility in a reducing environment but only if the solution was buffered.


We believe that the right combination of buffer, reducing media, and truncation size will allow us to create a functioning estrogen biosensor, but in the meantime we have a proof of principle for our biosensor platform in the form of a reducing environment sensor.

All of the composite parts submitted to the registry and experiments discussed on this wiki were created and performed by the 2011 iGEM team Berkeley undergraduates. Some of the basic parts (RBSes, etc.) were obtained through the Anderson lab, for which they have our thanks.