 "
"
Team:TecMonterrey/Project.html
From 2013.igem.org
KingDavid2 (Talk | contribs) |
KingDavid2 (Talk | contribs) |
||
| Line 605: | Line 605: | ||
<div class="menuhover">Teamm</div> | <div class="menuhover">Teamm</div> | ||
<img src="https://static.igem.org/mediawiki/2013/5/55/T_TecMonterrey.png" alt="team" class="buttoni"> | <img src="https://static.igem.org/mediawiki/2013/5/55/T_TecMonterrey.png" alt="team" class="buttoni"> | ||
| - | <a class="tag" href="./ | + | <a class="tag" href="./team.html">Team</a> |
<ul class="sub_navigation"> | <ul class="sub_navigation"> | ||
<li class="list"><a href="./team.html#MBRS">Members</a></li> | <li class="list"><a href="./team.html#MBRS">Members</a></li> | ||
Latest revision as of 03:55, 29 October 2013









Background

By harnessing the inherent ability of facultative anaerobic bacteria to colonize and grow in tumoral environments, this project aims to prove the functionality of four different modules that would work together as a bacterial cancer therapy using Escherichia coli as chasis: Toxicity module, Secretion module, Localized induction module, and Internalization module. The expression of tumor specific therapeutic proteins, Apoptin and TRAIL, conforms the toxicity module. For these proteins to have their effect they need to be located in the extracellular matrix, therefore we are developing a module with a secretion function using hemolysin secretory mechanism. The hypoxic microenvironment present in tumors can be used for the localized induction module of tumor specific proteins, using the promoters HIP and nirB. Finally, Apoptin needs mechanisms to enter tumor cells’ cytoplasm. Proteins with this requirement could reach the cytoplasm when coupled with the internalization module, resulting in a fusion with the TAT peptide.
Description
Tumor Specific Induction

An important part of this Project is the localized expression of the therapeutic proteins. For more than sixty years it has been known that some bacteria grow preferably in hypoxic conditions, being Escherichia coli one of these organisms. Tumors are known to be regions with low oxygen concentrations (hypoxic). With these factors in favor, three different hypoxia promoters were characterized.
HIP-1 promoter
Based on the work of Mengesha, et al. (2006), a hypoxia-inducible promoter (HIP-1) was used to control the expression of green fluorescent protein (GFP). This promoter was originally obtained from a portion of Salmonella pepT promoter with the combination of two binding sites for the FNR regulon and a TATAAT sequence.
Experimental work demonstrated that protein production was successful under acute and chronic hypoxia, but not under normoxia. To the correct operation of the promoter and all the anoxic metabolic pathways, there is one important gene: the fumarate and nitrate reductase (FNR). It is a transcriptional factor that inactives with the interaction of its sensory domain with oxygen molecules and it is present in Salmonella and E. Coli.
For this project it was designed a construct for the expression of GFP under hypoxic conditions to be used in BL21 E. coli strains. This constructs contains the FNR promoter and CDS native form E. coli, followed by a double terminator, the HIP-1 promoter designed by Mengesha, et al. (2006) and all the sequences required for the GFP expression. This expression cassete was cloned into a pUC57 plasmid.

nirB promoter
According to Peakman, et al. (1990) the nirB gene is the required for the synthesis of the nitrite reductase apoprotein and cysG proteins required for the sirohaem synthesis. This protestetic group is fundamental in the anoxic metabolic pathways of E. coli and it is used by many enzymes to complete a six-electron.
Reduction of sulfur and nitrogen. Knowing that these genes are specifically activated in E. coli during anoxic growth, in this project it was used the nirB promoter to make a cassette of expression to measure the effect of hypoxic environment over the expression of GFP.
The cassette is composed by: the NirB promoter (BBa_K905000 sequence reported in iGEM) and all the sequences necessary to express GFP, all cloned in pUC57 vector and transformed in BL21 E. strain.

Chimeric promoter
According to the fundaments of the Hip-1 construct and the nirB promoter, it was designed a new cassette controlled by those parts over the expression of GFP:

The BioBricks produced by the Hipoxia promoter module were:
BBa_K1166000 BBa_K1166001
BBa_K1166001

Therapeutic Proteins Production

The bacterial system expresses two therapeutic proteins and their fusions to a secretion sequence. The toxins used in this study were Apoptin and TRAIL, which are apoptosis-inducing proteins. The expression of these tumor specific therapeutic proteins conforms the toxicity module.
The constructs below are designed to express the therapeutic proteins by the use of IPTG.


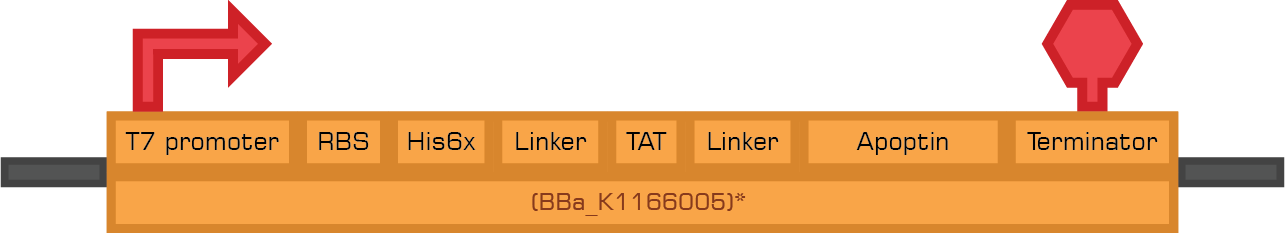

Based in the Therapeutic proteins module we designed two new biobricks for the registry:
Bba_K1166004 Bba_K1166005
Bba_K1166005

Apoptin
Apoptin, also called VP3, is a protein from the Chicken Anemia Virus (CAV) known to cause p53-independent apoptosis in more than 70 human cancer cell lines including hepatoma, osteosarcoma, melanoma, cholangiocarcinoma, colon carcinoma, lung cancer, breast cancer, prostate cancer, cervix cancer, gastric cancer while leaving normal cells unharmed. Apoptin contains a bipartite nuclear localization signal (NLS1: aa 82-88 and NLS2: aa 111-121) and a nuclear export signal (NES) (aa 97-105). In cancer cells, Apoptin localizes to the nucleus while in normal cells it's found in the cytoplasm; it is thought that the bipartite NLS is activated by phosphorylation at Threonine 108 which only happens in cancer cells.
Since Apoptin exerts its proapoptotic activity intracellularly, it is usually fused at the N-terminus with a cell-penetrating peptide such as the TAT peptide.
TRAIL
TRAIL (TNF Related Apoptosis Inducing Ligand) is known to cause apoptosis in various cancer cell lines with minimal cytotoxicity toward normal cells. TRAIL induces apoptosis by binding to two death receptor domains, TRAIL-R1(DR4) and TRAIL-R2(DR5). The binding of TRAIL triggers the trimerization of the death receptors that recruit and activate FADD a death domain-containing protein, FADD then recruits and activates caspase-8, leading to the formation of the death-inducing signaling complex (DISC).
Besides TRAIL-R1(DR4) and TRAIL-R2(DR5) TRAIL also binds to three antagonistic decoy receptors TRAIL-R3 (DcR1), TRAIL-R4 (DcR2) and osteoprotegerin, however, these receptors do not induce apoptosis but can protect cells from TRAIL-induced apoptosis.
Enhanced Secretion
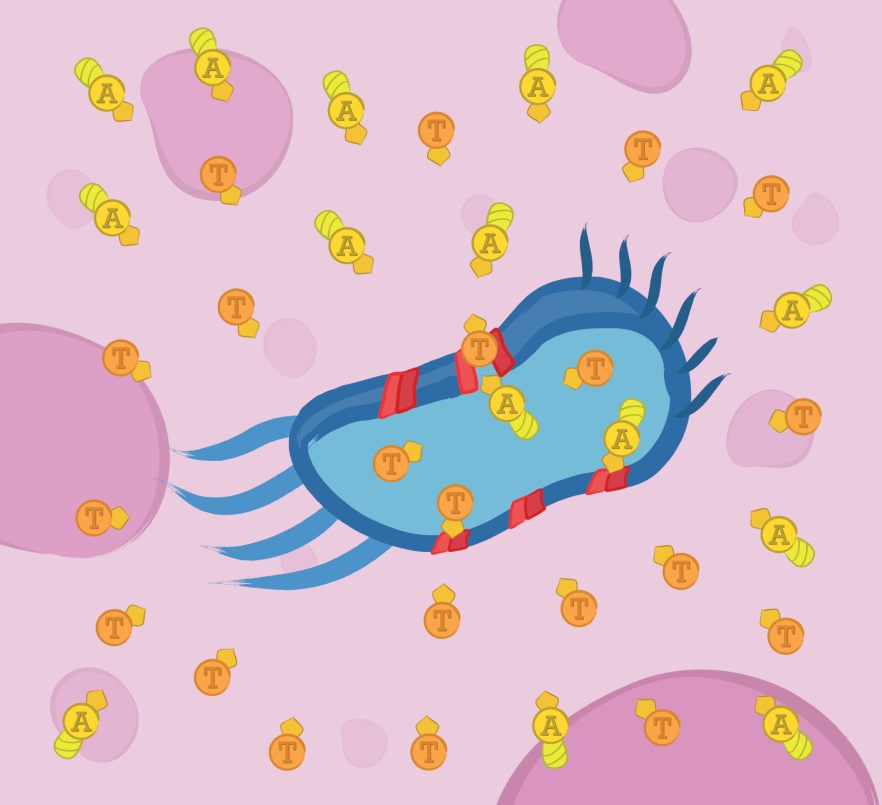
In order for TRAIL and TAT-Apoptin to exert their anticancer activity, they must reach the extracellular matrix in the first place. This module uses the type I a-hemolysin secretion system of Escherichia coli to achieve that feat.
The construct designs for this module were:
The constructs above, each contain a Histidine6x tag, allowing purification and further detection by western blot using a mouse monoclonal anti-his antibody.
The first two fused with secretion sequence HlyA, required to be exported to the medium, while the other two do not contain it, in order to serve as negative controls.


The last two constructs contain a GFP, the first one tagged with HlyA and the second one left untagged. In order to be measured for fluorescence in both the interior of the cell, and in the media.

The last construct is required to be co-transformed with all of the constructs already mentioned in the secretion module, in order to provide the required secretion proteins.
Based in the secretion module we designed a new Device for the registry:
BBa_K1166002
This is a device that allows a protein to be secreted by means of the alpha-hemolysin secretion system in E. coli. It’s designed so that the only thing that you have to do is to assemble your protein part in the device via the biofusion standard (BBF RFC 23), (Cut the device with EcoRI and XbaI, cut your protein part with EcoRI and SpeI, mix & ligate). This procedure will leave your part in frame with a signal peptide.
Note: It’s important that your part doesn’t contain a stop codon nor a terminator since your protein will be fused at the C-terminus.
In order for TRAIL and TAT-Apoptin to exert their anticancer activity they must reach the extracellular matrix in the first place. This module uses the type I alpha-hemolysin secretion system of E. coli to achieve that feat.
The mechanism of secretion is simple: In the cytosol, the substrate is recognized by means of a signal peptide and then translocated directly into the extracellular medium. The translocator consists of three proteins: HlyB, an ATP binding cassette; HlyD, a membrane fusion protein; and TolC, an outer membrane protein. Regarding the signal peptide, TAT-Apoptin and TRAIL were engineered to be fused C-terminally to the last 60 amino acids of HlyA (the natural substrate) since it has been shown that this C-terminal region of HlyA is both necessary and sufficient to direct secretion. While HlyB and HlyD are considered strain specific proteins, TolC is a component of multiple trans-membrane systems in many microorganisms, therefore we co-expressed only HlyB and HlyD.
Internalization
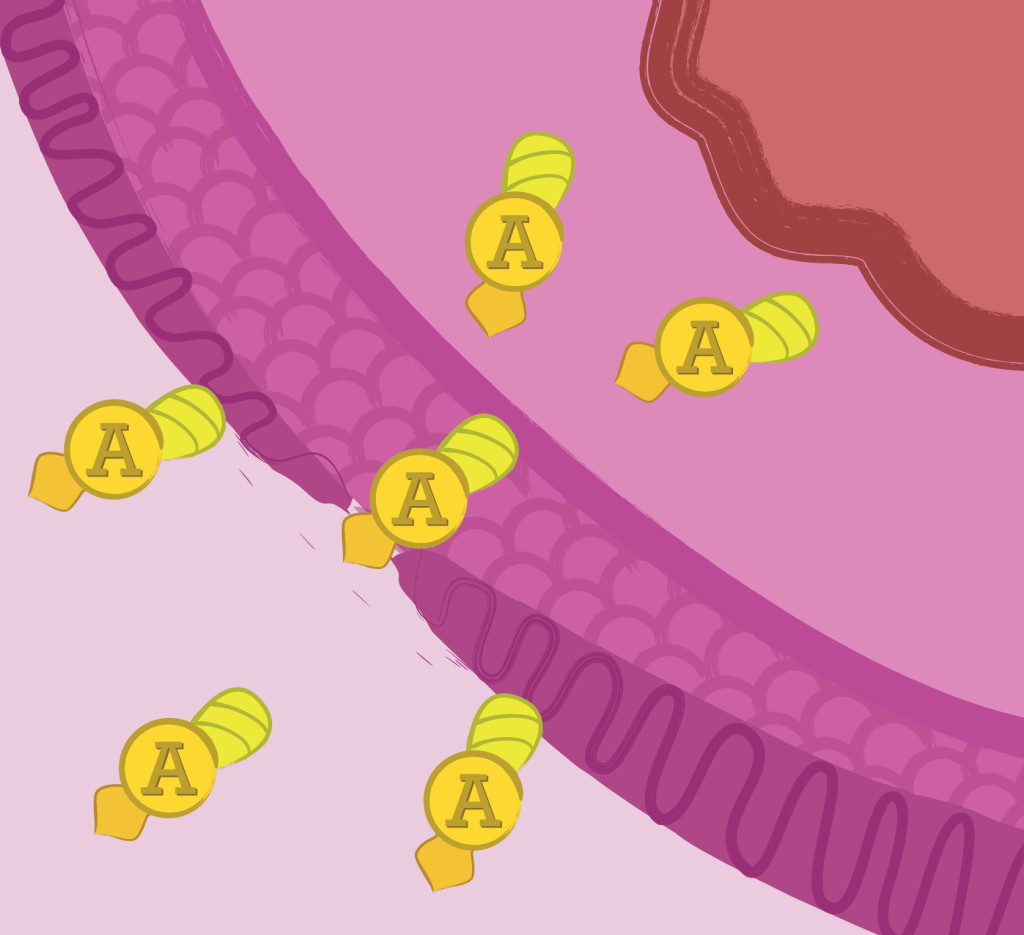
Internalization of Apoptin into the cytosol of human cells was confirmed using a TAT-GFP as a proof of concept approach.
The internalization module was characterized through the use of TAT-GFP fusion protein purified using His-tag affinity columns. Internalization essay was performed on grown human cells (NIH-T3 and CACO-2) treated with 1-10 µg/mL TAT-GFP followed by a Western Blot analysis of the lysates and a fluorescence essay of the supernatant in a 96-well microplate.
The constructs required for this module were:

The first constructs produces a GFP tagged with both a histidine6x tag and TAT internalization peptide.

The second construct produces a GFP tagged with only a histidine6x tag serving as a negative control for the internalization assay.
Based in the internalization module we designed a new biobrick:
BBa_K1166003The biobrick was designed in a way that the gene of interest is inserted in-frame at the 3’ end of TAT. This assures internalization of the protein of interest.

Internalization module relies on the ability of the HIV-1 trans-activator protein Tat transduction domain to deliver proteins across the cell membrane. Tat-fusion proteins internalization takes place in approximately 20 minutes and is dependent on the extracellular concentration of the Tat-fusion protein. (Vocero-Akbani et al., 1999).
In normal cells it stays in the cytoplasm where it has no significant effect, but in cancer cells it migrates into the nucleus where it triggers apoptosis. (Guelen, L. et al., 2004).
References
- Mengesha, A., Dubois, L., Lambin, P., Landuyt, W., Chiu, R., Wouters, B., & Theys, J. (2006). Development of a Flexible and Potent Hypoxia-Inducible Promoter for Tumor-Targeted Gene Expression in Attenuated Salmonella. Cancer Biology and Therapy, 1120-1128.
- Peakman, T., Crouzet, J., Mayaux, J., Busby, S., Mohan, S., Harborne, N., Cole, J. (1990). Nucleotide sequence, organisation and structural analysis of the products of genes in the nirB - cysG region of the Eschevichia coli K-12 chromosome. European Journal of Biochemistry Volume 191, Issue 2, 315-323.
- Unden, G., & Schirawski, J. (1997). The oxygen responsive transcriptional regulator FNR of Escherichia coli: The search for signals and reactions. Molecular Microbiology 25, 205-210.
- Silhol, M., Tyagi, M., Giacca, M., Lebleu, B. & Vivès, E. (2001). Different mechanisms for cellular internalization of the HIV-1 Tat-derived cell penetrating peptide and recombinant proteins fused to Tat. European journal of biochemistry / FEBS 269, 494–501.
- Guelen, L. et al. (2004). TAT-apoptin is efficiently delivered and induces apoptosis in cancer cells. Oncogene 23, 1153–65.
- Vocero-Akbani, A., Heyden, N., Lissy, N., Ratner, L. & Dowdy, S. Killing. (1998). HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein. Nature medicine 5, 29–33.
Modelling
Internalization Process

We’ve decided to make this deterministic math model because we considered that it will be useful for future applications in the analysis of similar biological systems (internalization processes).
Our deterministic math model is a mechanistic model following the bases of the Michaelis Menten kinematics. We decided to make a mechanistic model for the internalization process because the protein that is located outside the cytoplasm of the cancer cell (PA) will have an active transport through the cancer cell membrane. And according to Christopher S. Lobban (1994) the facilitated diffusion exhibits a Michaelis Menten kinetics. We can appreciate in our model that the PA will bound the MC2 , and it will form the enzyme-substrate complex MC2PA. This enzyme-substrate complex represents the way as PA will pass into the cytoplasm, as there is not an enzyme that can make reversible this process all the PA that is inside of the cytoplasm will have a higher concentration with the pass of the time. One thing that we considered was that in the interfacial enzymes of a membrane it is not appropriate to assume that the concentration of substrate is localized in certain places; For example, over the membrane or near to the enzymatic activity (Nate Cermak, 2009).
PC represents our protein inside the cytoplasm and we can say the there are two ways that the protein will get to be inside of the nucleus. If we see in the model one way is irreversible and the other is reversible. The one that is irreversible consists when the PC is phosphorylated and gets to pass inside the nucleus. As there are not an enzyme inside the nucleus that can make this process irreversible the concentration of the protein phosphorylated inside the nucleus will be higher with the pass of time (PFN). In the other way that is reversible, is when the protein in the cytoplasm (PC) is not phosphorylated and it enters the nucleus. Here there is an enzyme in the nuclear envelop that can bound the protein in the nucleus (PN) and make that it to pass again to the cytoplasm. This information was explained in the internalization protocol. And the model shows that the concentration of PFN will trend to increase, PC will reach a maximum, and the concentration of PA trend to decrease.
We proved this information by using Microsoft Excel. In the file that we made are included all the differential equations derived from the kinematics of the internalization process. The differential equations that we got were the following:

In summary, our model behaves as predicted, showing that a mechanistic model is appropriated to describe a system of internalization based on the mechanism of TAT-Apoptin internalization on mammalian cells and mammalian cancerous cells.
A further simulation of the concentration of the protein outside of the cell (PA) in the cytoplasm (PCT) and in the nucleus (PNT) shows that the protein tends to enter the nucleus at equilibrium, being slowed by the action of nuclear membrane secretion protein (MNS), which effect lowers together with the amount of non-phosphorylated protein.

References:
Lobban, Christopher (1994). Seaweed Ecology and Physiology. New York, USA: Cambridge University Press. Pág. 172
Cermak, N. (03 de 12 de 2009). Fundamentals of Enzyme Kinetics: Michaelis-Menten and Deviations. Recuperado el 10 de 09 de 2013, de http://cermak.scripts.mit.edu/papers/383final_cermak_enzymekinetics_20090312.pdf
Security and Safety Considerations
Since the designing phase of our project, we were aware that every project may have safety implications and that it is our responsibility to address them in an appropriate manner.
From the beginning of the brainstorming, the main idea involved the expression of therapeutic proteins that would act as toxins for tumor cells. At this point, we realized that we needed more specificity if healthy tissue was to be unharmed. In the hypothetic case that our bacteria were liberated into the environment and some way it survived and maintained its plasmid without a selectivity marker, it would be a catastrophe. This imaginary scenario led us to the idea of implementing safety locks in the design of our project.
Our first task to design our genes in a way their products wouldn’t harm us, was to think about which therapeutic proteins we were going to produce as tumor toxins. After bibliographic research we found out that the soluble part of the protein TRAIL (BBa_K1166004) and TAT-Apoptin (BBa_K1166005) were proteins that have antitumoral activity with high specificity leaving healthy tissue in its majority unharmed.
After we decided which proteins were going to be expressed, we also realized that its specificity could be enhanced if we managed to somehow produce them solely in the tumor. After weeks of bibliographic research, we learnt that in general, tumors exhibit an environment with a lower amount of oxygen concentration (hypoxia). From this research, we decided the regulation for our project to be controlled by a hypoxic induction promoter, for which we created FNR hypoxic regulation part (BBa_K1166001) and a new hypoxic promoter HIP-1 (BBa_K1166000), along with the characterization of an existing hypoxia promoter (BBa_K905000) complemented with our FNR regulation part.
We believe that these precautions along with good practices at the execution of the project will work to maintain and highlight safety in our lab and for our society.
Also, during the execution phase of our project we followed the top security requirements for the materials we were using. First of all, it is important to cover a brief description of both the materials used in the project, and the organisms employed and their characteristics. It is important to understand that we are always exposed to risk and no experiment is 100% risk free. Nevertheless, our job is to minimize the probability of being exposed to any danger.
During the realization of the project we did the following procedures:
- Miniprep (plasmidic DNA extraction)
- Agarose gel DNA electroforesis
- Restriction enzyme analysis
- Escherichia coli Transformation by CaCl2
- Inducible promoter induction with L-Arabinose, IPTG and hypoxia
- SDS-PAGE
- Western Blot
- Cell lysis and Protein Extraction
- Mammalian cells toxicity/internalization assay
The realization of the named protocols implies the use of some hazardous materials. These substances require special treatment and disposal. In our protocols we use the following dangerous substances:
- Acrylamide
- Ethidium Bromide
- Acetic acid
- Chlorine
- HCl
- NaAC
- NaOH
- ß-Mercaptoethanol
- SDS
- Coomassie Blue
For us to use this kind of substances some security protocols are needed to be followed.
- Miniprep (plasmidic DNA extraction) uses solutions with EDTA, these solutions are made with nitrile gloves to prevent any possible contact with the skin, as it is known that EDTA has suspected effects on the reproductive system. Also solutions using NaOH and NaAc are made under a laminar flow hood to prevent any kind of dangerous inhalation.
- During the Agarose gel electrophoresis the dangerous part come when the researcher has to use Ethidium Bromide to reveal the gel. Ethidium Bromide intercalates into the DNA double strand, as a strong intercalator is mutagenic and carcinogen. To work with Ethidium Bromide, it is needed to designate a special workplace; also you need to restrict the number of people to the minimum. To keep the biosafety we followed all of the recommendations, also we used nitrile gloves that are less permeable to EtBr than latex gloves. Finally, we designated a special disposal bin for all EtBr–related residues.
- In the E. coli Transformation by CaCl2 protocol, we are exposed to the E. coli non-transformed strains. To prevent any escape to the environment of these strains, we perform every transformation under a biosecurity chamber. Any residues generated in this protocol that had contact with biological material are then disposed in a biohazard bag that is sealed and autoclaved in 121°C and 15psi for 15 minutes.
- Induction protocols consist in adding a specific substance to a cell culture for it to start the production of a specific protein. In our case, our inductors are L-Arabinose, IPTG and hypoxia. To prevent any damage by inhalation or any possible fire, the preparation of methanol is done in a gas extraction cabinet. Also, its sterilization is done by filtration to prevent any fire or explosion.
- SDS-PAGE is very usefull tool at the time when we need to make a protein analysis by size, nevertheless it is full of hazardous materials. To begin with the SDS-PAGE, you need to prepare the polyacrylamide gel. This is done by adding TEMED and PSA to Acrylamide. TEMED and PSA are harmful for the respiratory tract, so for its use we need to have precaution. Acrylamide is known for being a strong neurotoxin, so nitrile gloves are used. It is important to assign a separate special workspace for any procedure using these substances.
To prepare the protein sample, B-mercaptoethanol and SDS are needed, and both have toxic effects, especially B-mercaptoethanol. To keep our biosafety standards, we minimize the time of exposure to any of this materials, also we work using always nitrile gloves, and in our designated space.
After the preparation of the SDS-PAGE and the running of the gel, you need to stain with Coomassie Blue. As some of the acrylamide may have not polymerized yet, we keep our gloves on, especially because of the Coomassie staining. Coomassie Blue is a known flammable toxin that targets some of our organs.
For the Coomassie Blue to stain with more definition it is recommended to use a “fixating solution”, which contains methanol, acetic acid and water. For the preparation of this solution, an air-extraction cabinet is needed. Finally, we dispose of all the generated wastes in a bin we labeled “PAGE wastes”, separated from our normal wastes.
An iGEM guide for evaluating the commercial potential and profitability of a technology project using the Quicklook Methodology.
We have noticed that iGEM teams throughout the years have developed and are developing novel projects. However, in most of the cases they only stay in the stage of investigation and results but never go further. We believe that many of these projects could have an important impact in the community and in society if they go to the business world by commercialization of the technology.
We saw this problematic and start investigating ways to commercialize a project, we found out a new methodology called Quicklook. We believe that with this tool other iGEM teams will be able to evaluate and create and idea of how valuable and profitable their research is in order to commercialize their idea and contribute to what biobusiness is. The description of what Quicklook Methodology is and the essential parts of how to make one are described below.
What is a Quicklook?
It is a methodology developed by the IC2 institute of the University of Texas to evaluate the potential commercial interest of an idea, area of research or technology. This methodology helps the research team to evaluate two important risks:
- Technological Risks: Will the technology work?
- Market Risks: Who will purchase the technology?
The QuickLoook® Methodology consists of seven steps:
- Identify and analyze Potential Markets
- Identify competition, end users, distributors and potential licensees
- Search patents for similar technologies.
- Identify the resources a company would need to enter the market with the technology analyzed.
- Identify potential risks and plan how to overcome them.
- Contact experts and ask for their opinion.
- Write the final report.
A Quicklook analyses aspects like technology, benefits and opportunity, market interests, potential markets, technology phases, patent stage, threats of new entrants and of existing competition and opportunities to get into the market
How to make a Quicklook
There are several steps that have to be done in order to have a Quicklook report.
- Technology description: in simple terms the technology develop is described by the creator. Some questions should be answer in this section.
- What does the technology/ research/ system do?
- What problem does it solves?
- How does it work.
- Intellectual property.
- What kind of protection does your IP have?
- What other IP that are alike to your technology have you explored?
- Examinate competitors and competing technologies.
- Who is your competition
- What is the difference between your competition and your technology?
- How has the market accepted the competition technology?
- What are the strengths and the debilities of the competition´s technology?
- Identify and explore market commercial potential
- Who will use your technology?
- Who needs it?
- In what industry will it be used?
- Define your market; size, structure.
- Primary marketing research
- Interviews with experts in the area like academics.
- Interview with your potential market
- Secondary marketing research
- Collect existent information about your technology.
- Process the information in understandable specific data.
- Get information about the competition.
- Obtain historical and quantitative data of the target market.
- Quicklook report
- Write all the information above in a report including all the aspects in the picture above.

Here is the link to download our team iGem TecMonterrey short Quicklook Metodology about our project research.
Patents
What is a patent?
What is a patent?
A patent is a set of exclusive rights given to an inventor by a government for a limited time period in exchange for reveling to the general public the invention. This legal document grants the exclusivity of making, using or selling an invention or products made by an invented process.
What is patentable?
For an invention to be patentable it should be something new, it must involve an inventive step or it shouldn’t be obvious for someone with knowledge in the subject, and capable of industrial application. An invention is considered to be new if it is not known or used in a public way in any part of the world.
How to know if an invention is patentable?
Search the different patent office databases for similar inventions to the one developed. Recommended databases are the US Patent Office, European Patent office, and the office of the country where the invention is being or was developed. Within each patent office search for granted patents and patent applications.
Within each patent application there’s a section called Claims. The claims of a patent define in technical terms the extent of what is protected by the patent. This section should by analyzed and compared to the invention that is intended to be patented.
Previous Release of an Invention
Article 18 of the Mexican Law of Industrial Property:
The public release of an invention will not affect its novelty if it was made during the previous twelve months of the patent filling date. When presenting the patent application the applicant should specify the public release of the invention in the patent application form.
Patent Application
A patent application consists of a description of the invention called the patent specifications and other forms. The patent document consists of several sections.
- Description or Patent Specification.
- It generally contains the background and overview of the invention as well as a detailed description of it.
- The Article 47 of the Mexican Law of Industrial Property states that a patent specification should be perfectly clear so any one with middle knowledge in the subject could replicate the invention.
- Patent Claims.
- This section, as mentioned previously, defines the scope of protection of a patent.
- Filling Dates.
- This is the date at which the patent form was filed in the patent office.
- Patent Summary.
- It’s a brief description of the technical development of the invention. It should be no longer than 200 words.
- Drawings.
- This section’s purpose is supporting the patent description.
References
Cámara de Diputados del H, Congreso de la Unión. (1991). Ley de la Propiedad Industrial. Diario Oficial de la Federación.
Instituto Mexicano de la Propiedad Intelectual. (2012). Guía del Usuario Patentes y Modelos de Utilidad. México: Instituto Mexicano de la Propiedad Intelectual.
Region. (n.d.). Technology Assessment Training - Global Commercialization Group. IC² Institute, the University of Texas at Austin | Innovation, Creativity, Capital. Retrieved September 27, 2013, from http://www.ic2.utexas.edu/global/services/education/assessment/
Region. (n.d.). Technology Assessments - Global Commercialization Group. IC² Institute, the University of Texas at Austin | Innovation, Creativity, Capital. Retrieved September 27, 2013, from http://www.ic2.utexas.edu/global/services/assessments/
Future work
Validation by experts
An important aspect of every project is its validation by experts, although a project may seem perfect for its creators there could be hidden issues. Additionally, this validation can greatly increase the possibilities of funding your project.
With this in mind, we took on the task of looking for experts that could gave us feedback about our project.
Here's the list of Drs. that we visited:
- Dr. Luis Mario Villela - Escuela de Medicina y Ciencias de la Salud
- Dr. Adolfo Isassi Chapa - San Jose Tec de Monterrey Hospital
- Dr. Servando Cardona Huerta - San Jose Tec de Monterrey Hospital
- Dr. Jorge Martinez - San Jose Tec de Monterrey Hospital
- Dr. Cesar Gonzalez de Leon - Opcion/ Muguerza
- Dr. Augusto Rojas Martinez - Universidad Autonoma de Nuevo Leon
- Dr. Alfonso Duenas Gonzalez - Instituto Nacional de Cancerologia
We got invaluable feedback, particulary something that many Drs. pointed out was the sepsis issue. They thought it may be a better idea an intratumoral injection instead of an intravenous (i.v.) therapy, this with the goal of focalizing the inoculum and therefore reduce the interaction of the bacteria with the immune system.
We also thought of some approaches that may be used in combination to tackle this problem.
Anti-LPS peptide
Lipopolysaccharide (LPS) is a ubiquitous cell surface component of E. coli and also one of the major toxins responsible for initiating sepsis (Lynn et al, 1992). Interestingly, it has been shown that the interaction of LPS with the immune system is mediated by a protein called LPS binding protein (LBP)(Jiang et al, 2000). Therefore, a way to reduce the risk of sepsis would be to block this interaction. It turns out that there are some molecules known to achieve that effect such as the Limulus anti-LPS factor (LALF), a small basic peptide found in hemocytes of the marine chelicerates Tachypleus tridentatus and Limulus polyphemus. An approach could be the administration of LALF during the therapy.
Oral therapy
The idea of administering bacteria orally in an effort to fight cancer already has been explored. It has been shown that Bifidobacterium breve fed to mice were detected specifically in tumors at levels similar to i.v. administration. (Cronin et al, 2010). This was posible because some bacteria are able to translocate the gastrointestinal tract (GIT) including E. coli (Mahjoub-Messai et al, 2011).
Also, we thought that some additional elements must be included in our therapy:
Plasmid loss prevention
It's well known that in absence of a selective pressure a plasmid can be lost if it doesn't confer an advantageous trait. Inside the tumor where no antibiotic is found it's critical to use a strategy to avoid this problem since plasmid loss will be reflected in loss of dosage of therapeutic proteins. A simple strategy would be an auxotrophic marker.
Horizontal gene transfer prevention
This issue already has been addressed on iGEM. A strategy could be the use of a toxin/anti-toxin system such as the Endolysin-Holin system proposed by the Imperical College of London.
References
- Lynn, W.A., and D.T. Golenbock. (1992). Lipopolysaccharide antagonists. Immunol. Today. 13(7):271–276.
- Qingqi Jiang, Sachiko Akashi, Kensuke Miyake and Howard R. Petty. (2000). Cutting Edge: Lipopolysaccharide Induces Physical Proximity Between CD14 and Toll-Like Receptor 4 (TLR4) Prior to Nuclear Translocation of NF-?B B. J.. Immunol. 165:3541-3544.
- Cronin M, Morrissey D, Rajendran S, El Mashad SM, van Sinderen D, O'Sullivan GC, Tangney M. (2010). Orally administered bifidobacteria as vehicles for delivery of agents to systemic tumors. Mol Ther. 18(7):1397-407.
- Mahjoub-Messai F, Bidet P, Caro V, Diancourt L, Biran V, Aujard Y, Bingen E, Bonacorsi S. (2011). Escherichia coli isolates causing bacteremia via gut translocation and urinary tract infection in young infants exhibit different virulence genotypes. J Infect Dis. 203(12):1844-9.