 "
"
Team:UChicago/Notebook
From 2013.igem.org
(→Tuesday, August 20, 2013) |
(→Tuesday, August 20, 2013) |
||
| Line 262: | Line 262: | ||
|5.5 | |5.5 | ||
|} | |} | ||
| - | + | used 18E (e. coli promoter from plate 5) instead of pSB1A3, has plasmid [http://parts.igem.org/File:PBca1020-r0040.jpg BBa_J61002] w/ AmpR | |
| + | |||
*The concentrations of the components of the ligations were as follows: | *The concentrations of the components of the ligations were as follows: | ||
| Line 306: | Line 307: | ||
|} | |} | ||
| - | *Digested kerA overnight (Ethan) | + | *Digested ([[https://2013.igem.org/Team:UChicago/Protocols#Enzymatic_digestion Enzymatic Digestion]])kerA overnight (Ethan) |
**10 ul kerA | **10 ul kerA | ||
**2 ul digest buffer | **2 ul digest buffer | ||
| Line 319: | Line 320: | ||
**3 ul H2O | **3 ul H2O | ||
| - | [[https://2013.igem.org/Team:UChicago/Protocols# | + | |
| + | ====Wednesday, August 21, 2013==== | ||
| + | ---- | ||
| + | '''Transformation with kerA-pSB1A3 with control experiments and Transformation of B. subtilis WB700 with pUB110''' | ||
| + | |||
| + | *Transformed E. coli ([[https://2013.igem.org/Team:UChicago/Protocols#Ivan.E2.80.99s_protocol_for_his_competent_cells_in_PCR_tubes: Transformation protocol]]) with yesterday’s kerA-pSB1A3 ligation | ||
| + | **used 8/19 kerA digest (90ng/ul), the only pSB1A3 digest (24ng/ul), and 18E miniprep (963ng/ul) | ||
| + | **incubated overnight at 37C starting 2:30pm | ||
| + | **the following experiments, including controls, were performed | ||
| + | {| class="wikitable" | ||
| + | |'''plate/tube #''' | ||
| + | |1 | ||
| + | |2 | ||
| + | |3 | ||
| + | |4 (has small crack) | ||
| + | |5 | ||
| + | |- | ||
| + | |tube | ||
| + | |3:1 | ||
| + | |5:1 | ||
| + | |10:1 | ||
| + | |cut vector w/ ligase - '''control''' | ||
| + | |uncut vector w/o ligase - '''control''' | ||
| + | |- | ||
| + | |Insert (kerA digest 8/19) | ||
| + | |1ul | ||
| + | |1.5ul | ||
| + | |3ul | ||
| + | |N/A | ||
| + | |N/A | ||
| + | |- | ||
| + | |Vector (pSB1A3 digest) | ||
| + | |1ul | ||
| + | |1ul | ||
| + | |1ul | ||
| + | |1ul | ||
| + | |(18E)** 1ul | ||
| + | |- | ||
| + | |Buffer | ||
| + | |1ul | ||
| + | |1ul | ||
| + | |1ul | ||
| + | |1ul | ||
| + | |1ul | ||
| + | |- | ||
| + | |Ligase | ||
| + | |0.5ul | ||
| + | |0.5ul | ||
| + | |0.5ul | ||
| + | |0.5ul | ||
| + | |0.5ul | ||
| + | |- | ||
| + | |H2O | ||
| + | |6.5ul | ||
| + | |6ul | ||
| + | |5.5ul | ||
| + | |7.5ul | ||
| + | |8.5ul | ||
| + | |} | ||
| + | used 18E (e. coli promoter from plate 5) instead of pSB1A3, has plasmid [http://parts.igem.org/File:PBca1020-r0040.jpg BBa_J61002] w/ AmpR | ||
| + | |||
| + | |||
| + | *About control experiments: | ||
| + | **'''Uncut vector (BBa_J61002) withOUT ligase''': checks for viability of competent cells and antibiotic resistance of the plasmid, as well as transformation procedure itself | ||
| + | ***expect colonies | ||
| + | ***since we are using a linearized plasmid, is there another uncut vector we could use that has the same antibiotic resistance so we can check the competency of our cells? | ||
| + | **'''cut vector WITH ligase''': shows background due to vector recircularization (and uncut vector) | ||
| + | ***expect few colonies (few more than without ligase), since only the plasmids which did not get cut during the digest or the plasmids which did get cut but are able to recircularize with the ligase will be transformed | ||
| + | ***this is the most important control. theoretically, there should be no colonies, but there is always some background, so as long as we have a lot more colonies on the vector with insert (actual ligation reaction) plate, it's okay. | ||
| + | *The following controls were not carried out but should be and are useful for future experiments: | ||
| + | **'''cut vector withOUT ligase''': shows background due to uncut vector | ||
| + | ***expect few colonies, since only the plasmids which did not get cut during the digest will be transformed | ||
| + | ***only do this one if you have extra cut vector, if we don't set it up it's not a big deal, since cut vector with ligase will just show the total background due to uncut vector and vector recircularization. | ||
| + | **'''insert ONLY WITH ligase''': checks for contamination of intact plasmid in ligation or transformation reagents | ||
| + | ***expect no colonies, since if there are colonies it indicates contamination of intact plasmid carrying antibiotic resistance in reagents since only the insert was used which should not be able to recircularize into a construct which can be transformed | ||
| + | ***this control is optional, I never do it, but if we are concerned about contamination of reagents with plasmids this could help, so I think we should do it if there is extra insert | ||
| + | **More info on control ligation experiments can be found [http://www.addgene.org/plasmid_protocols/DNA_ligation/ here] | ||
===Week 4=== | ===Week 4=== | ||
Revision as of 19:07, 26 September 2013
| Protocols |
|---|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Lab Notebook
Contents |
June
Week 3
Tuesday, June 18, 2013
First iGEM Meeting of the Summer!
- All student members gathered in the lab.
- We toured the lab, prep room, and incubator room, and we discussed the lab rules.
- We also split into teams to discuss lab logistics: contacting funding sources, ordering reagents/materials, compiling protocols, recording our electronic lab notebook, and making plates/media.
Week 4
Wednesday, June 26, 2013
Planning Wet Lab Work
- We split into planning sub-teams to plan out different parts of the project with protocols, timelines, and ordering reagents.
- Plasmid Design
- Media and Competent Cells
- E. coli and B. subtilis Transformation
- Keratinase Assay and His tagging
- Running DNA gels, SDS-PAGE gels, and western blots
- We will also be holding weekly meetings with our graduate student advisers and biweekly meetings with our faculty advisers to provide updates.
- Wet lab work begins tomorrow!
Friday, June 28, 2013
Researching the Sequence of kerA
- Planning subteams continued to order reagents and compile protocols.
- The plasmid design team decided on gibson assembly to produce the kerA biobrick with geneblocks ordered from IDT.
- kerA sequence was obtained from “Nucleotide Sequence and Expression of kerA, the Gene Encoding a Keratinolytic Protease of Bacillus licheniformis PWD-1” (Lin et al. 1995).

- Two putative promoters (red), a possible ribosomal binding site (green), and a transcriptional terminator sequence (light blue, single underline) are indicated. The putative starting residues of the preprotein (pre = blue), proprotein (pro = pink), and mature protein (mature = purple) are indicated.
- EcoRI (orange) and PstI (blue) sites within the gene are indicated and must be removed to produce the kerA biobrick.
July
Week 1
Wednesday, July 3, 2013
BioBrick Design Presentation and Grad Adviser Meeting
- Designed a biobrick with:
- C terminus His-tagged kerA
- CATCATCATCATCATCAT
- Prefix: gaattcgcggccgcttctagag
- Suffix: tactagtagcggccgctgcag
- C terminus His-tagged kerA
- Moreover, the bio brick must include
- Eliminated EcoRI and PstI restriction sites from kerA nucleotide seq
- EcoRI (gaattc--814) and PstI (ctgcag--978)
- Conserved aa seq + biobrick prefix + suffix
- Secretion signal peptide
- Inducible promoter
- Eliminated EcoRI and PstI restriction sites from kerA nucleotide seq
- We also presented a feasible timeline of our project, which included:
- Assembly of biobrick and transformation into E. coli
- Transformation of B. subtilis
- Keratinase isolation and keratinase assays
- kerA mutagenesis for optimization of the keratinase
- We also split up basic lab duties!
- Ivan & Ethan are in charge of the IBC protocol.
- We can’t start wet lab work until our IBC protocol is finished!
- Ethan is in charge of minutes.
- Susan is in charge of ordering invoices.
- Annie is in charge of emailing advisors and members to plan meetings
- Chloe is getting the B. subtilis gene that expressing keratinase.
- Ivan & Ethan are in charge of the IBC protocol.
Thursday, July 4, 2013
Milk Agar Plates
- The purpose of milk agar plates is often to detect protease activity, including the activity of keratinases.
- A measurement of any baseline protease activity of our parent B. subtilis strain (WB700), which does not endogenously express any keratinases, is desired.
- Excess protease activity of our parent B. subtilis strain may prevent our use of milk agar plates as an assay for keratinase expression of our future transformants with the kerA gene.
- Therefore, the parent B. subitilis strain (WB700) was streaked on the milk agar plates to observe baseline protease activity that could prevent us from detecting keratinase expression after transformation.
- [Milk Agar Plates] (in LB):
- Ideally, there should be a minimal zone of clearing around our B. subtilis colonies.
- Next Steps:
- Observe the results of our milk agar plates with B. subtilis colonies
Friday, July 5, 2013
Primer Design for kerA biobrick
- Designed primers for directed mutagenesis of our kerA biobrick using PrimerX:
- Primer pair 1 (EcoRI mutation, aat --> aac, asparagine)
- a. Forward: 5' GGTTAAAGTACTGAACTCAAGCGGAAGCGG 3'
- b. Reverse: 5' CCGCTTCCGCTTGAGTTCAGTACTTTAACC 3'
- Primer pair 2 (Pst1 mutation, gca-->gcg, alanine)
- a. Forward: 5' GTTGTCGTTGTAGCTGCGGCAGGGAACAGCGGATC 3'
- b. Reverse: 5' GATCCGCTGTTCCCTGCCGCAGCTACAACGACAAC 3'
- Used APE for checking codons and mutating restriction sites
Saturday, July 6, 2013
Primer Design Part II
- Designed primers to isolate non-coding and coding sequence of kerA and add prefix and suffix
- forward primer + xbaI site:
- 5' TATCTAGAGGTCTATTCATACTTTCGAA 3'
- reverse primer + speI site (reverse complement):
- 5' ATTGATCATTGGACAACCTTCATCAG 3'
- We still need to design additional primers that will include the secretion signal peptide (in case it needs to be secreted out of B. subtilis) and kerA (From start to stop codon).
- These primers should include igem biobrick prefix and his tag (before stop codon) + suffix.
Week 2
Monday, July 8, 2013
Milk Agar Plate Results
- The results of our milk agar assay from 7/04/13 of the non-transformed B. subtilis strain WB700:
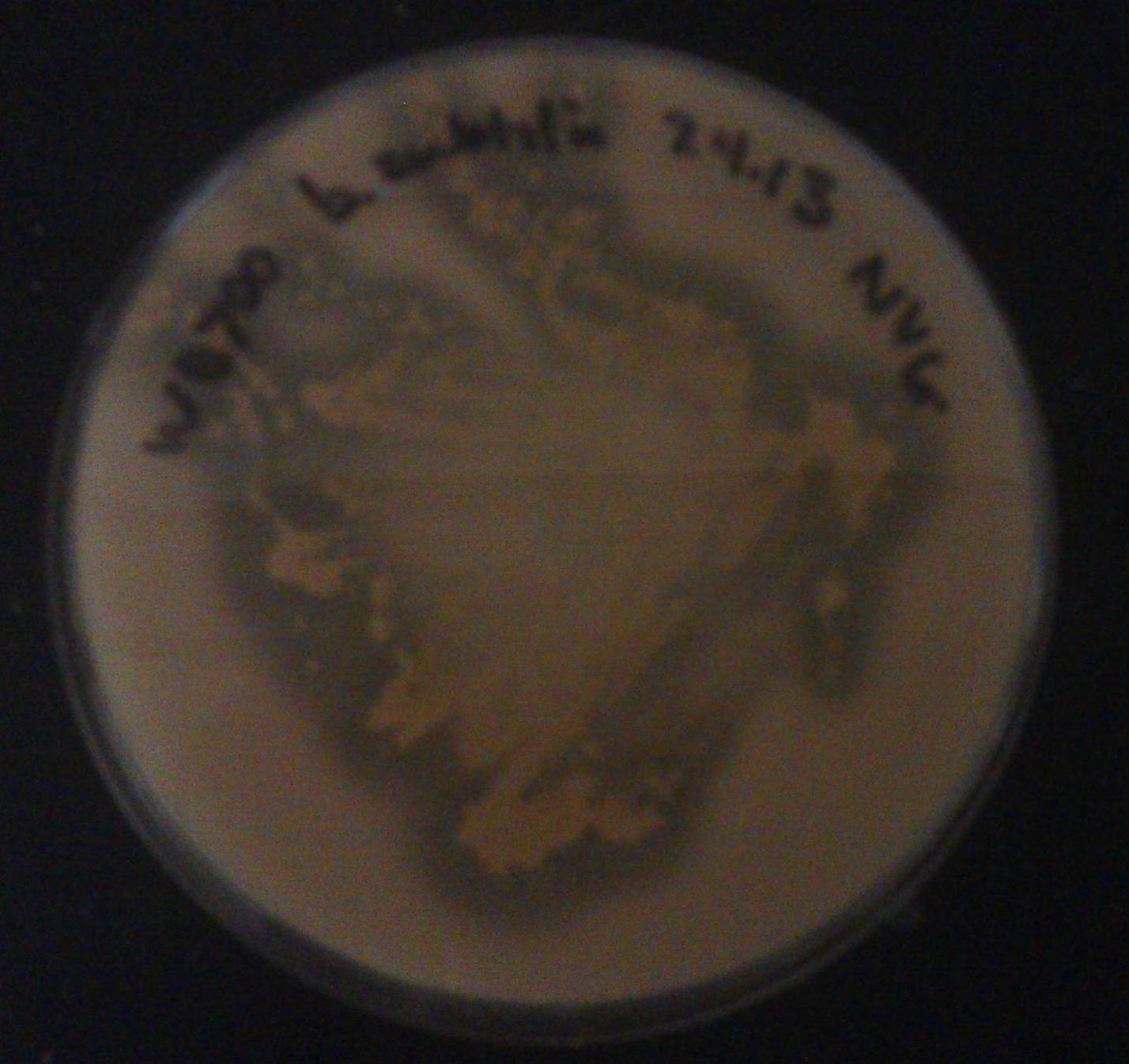
- The zone of clearing around the B. subtilis colonies indicates that our parent strain breaks down milk. Its prominent endogenous protease activity prevents us from using the milk agar assay as a screen for successfully transformed kerA-expressing B. subtilis.
- Alternative method to screen for transformed B. subtilis needed:
- The feather assay used in Saha and Dhanasekaran’s 2010 paper, “Isolation and Screening of Keratinolytic Actinobacteria form Keratin Waste Dumped Soil in Tiruchirappalli and Nammakkal, Tamil Nadu, India”
- Quantitative assay using azokeratin
Our future plans:
- For our promoter for kerA, Karyl, one of our graduate advisors, recommended P43, a constitutive promoter, to transform into B. subtilis.
- We also decided to use pUB110, a high copy number plasmid used in S. aureus and B. subtilis, to introduce kerA into our parent B. subtilis strain.
- pUB110 will also be made into a biobrick.
- We have to choose a suitable plasmid (either pSB1A3, pSB1C3, pSB1T3, or pSB1K3.m1) for our E. coli transformations.
Wednesday, July 10, 2013
Grad Advisor Meeting on further developed Keratinase Project
- We presented the primers we designed to our advisers, who suggested we double check them for self dimerization and 3’ overlaps with:
- We discovered that our forward primer forms a self-dimer and must be redesigned.
- We also need lysozyme to digest the peptidoglycan of B. subtilis in order to miniprep it.
- We decided to use the iGEM plasmid pSB1K3.m1 for our E. coli transformations.
- To screen for keratinase activity, instead of using the feather assay, which takes up to a month for visible results to appear, we decided to use the azokeratin assay as the secondary screening method.
Week 3
Week 4
August
Week 1
Week 2
Week 3
Tuesday, August 20, 2013
kerA-pSB1A3 ligation and RFP digestion results
- Measured the concentration of the RFP digested yesterday
- [8/19 RFP Digest] = 74.7 ug/ul
- Ran a 2% gel of the RFP digest
- Lane 2: Ladder (8 ul)
- Lane 4: 8/19 RFP mp#2 Digest (2 ul) + loading dye (2 ul)
- Added 5/10 ul EtBr to the buffer
INSERT 8/20 GEL PIC HERE
- There is only 1 band in lane 4.
- Email iGEM to request new Pveg+RBS construct purchased
- Ligated kerA and pSB1A3 with control experiments (Ivan)
- The ligations were set up as follows:
| uncut w/o ligase Control | cut w/ ligase Control | 3:1 | 5:1 | 10:1 | |
| Vol. Insert kerA digest 8/19 (uL) | N/A | N/A | 1 | 1.5 | 3 |
| Vol. Vector pSB1A3 digest (uL) | (18E)** 1 | 1 | 1 | 1 | 1 |
| Vol. Buffer (uL) | 1 | 1 | 1 | 1 | 1 |
| Vol. Ligase (uL) | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Vol. H2O (uL) | 8.5 | 7.5 | 6.5 | 6 | 5.5 |
used 18E (e. coli promoter from plate 5) instead of pSB1A3, has plasmid BBa_J61002 w/ AmpR
- The concentrations of the components of the ligations were as follows:
- kerA 8/19 digest: 90ng/ul
- pSB1A3 digest: 24ng/ul
- 18E miniprep: 963ng/ul
- The ligations incubated at RT overnight
- Another kerA-pSB1A3 ligation is being run in parallel in case the first does not work (Ethan):
- Incubated overnight at 4°C
- ligation reactions:
- ker A gel extraction (8/16) = 115 ng/ul (~1 kb)
- ker A gel extract (8/13) = 75 ng/ul
- pSB1A3 digest = 25 ng/ul (~ 2 kb)
| reaction | 1 | 2 |
| kerA | 1 ul kerA (8/16) = 115 ng | 2 ul kerA (8/13) = 150 ng |
| pSB1A3 | 1 ul pSB1A3 = 25 ng | 0.5 ul pSB1A3 = 12.5 ng |
| molar ratio (insert/vector) | 2.3 | 6 |
| 1 ul 10X ligation buffer | 1 ul 10X ligation buffer | |
| 0.5 ul ligase | 0.5 ul ligase | |
| 6.5 ul water | 6 ul water |
- Digested ([Enzymatic Digestion])kerA overnight (Ethan)
- 10 ul kerA
- 2 ul digest buffer
- 0.5 ul EcoRI (F.D.)
0.5 ul SpeI (F.D.) 7 ul H2O
- Digested pSB1A3 overnight (Ethan)
- 5 ul pSB1A3 (linear)
- 1 ul digest buffer
- 0.5 ul EcoRI (F.D.)
- 0.5 ul SpeI (F.D.)
- 3 ul H2O
Wednesday, August 21, 2013
Transformation with kerA-pSB1A3 with control experiments and Transformation of B. subtilis WB700 with pUB110
- Transformed E. coli ([Transformation protocol]) with yesterday’s kerA-pSB1A3 ligation
- used 8/19 kerA digest (90ng/ul), the only pSB1A3 digest (24ng/ul), and 18E miniprep (963ng/ul)
- incubated overnight at 37C starting 2:30pm
- the following experiments, including controls, were performed
| plate/tube # | 1 | 2 | 3 | 4 (has small crack) | 5 |
| tube | 3:1 | 5:1 | 10:1 | cut vector w/ ligase - control | uncut vector w/o ligase - control |
| Insert (kerA digest 8/19) | 1ul | 1.5ul | 3ul | N/A | N/A |
| Vector (pSB1A3 digest) | 1ul | 1ul | 1ul | 1ul | (18E)** 1ul |
| Buffer | 1ul | 1ul | 1ul | 1ul | 1ul |
| Ligase | 0.5ul | 0.5ul | 0.5ul | 0.5ul | 0.5ul |
| H2O | 6.5ul | 6ul | 5.5ul | 7.5ul | 8.5ul |
used 18E (e. coli promoter from plate 5) instead of pSB1A3, has plasmid BBa_J61002 w/ AmpR
- About control experiments:
- Uncut vector (BBa_J61002) withOUT ligase: checks for viability of competent cells and antibiotic resistance of the plasmid, as well as transformation procedure itself
- expect colonies
- since we are using a linearized plasmid, is there another uncut vector we could use that has the same antibiotic resistance so we can check the competency of our cells?
- cut vector WITH ligase: shows background due to vector recircularization (and uncut vector)
- expect few colonies (few more than without ligase), since only the plasmids which did not get cut during the digest or the plasmids which did get cut but are able to recircularize with the ligase will be transformed
- this is the most important control. theoretically, there should be no colonies, but there is always some background, so as long as we have a lot more colonies on the vector with insert (actual ligation reaction) plate, it's okay.
- Uncut vector (BBa_J61002) withOUT ligase: checks for viability of competent cells and antibiotic resistance of the plasmid, as well as transformation procedure itself
- The following controls were not carried out but should be and are useful for future experiments:
- cut vector withOUT ligase: shows background due to uncut vector
- expect few colonies, since only the plasmids which did not get cut during the digest will be transformed
- only do this one if you have extra cut vector, if we don't set it up it's not a big deal, since cut vector with ligase will just show the total background due to uncut vector and vector recircularization.
- insert ONLY WITH ligase: checks for contamination of intact plasmid in ligation or transformation reagents
- expect no colonies, since if there are colonies it indicates contamination of intact plasmid carrying antibiotic resistance in reagents since only the insert was used which should not be able to recircularize into a construct which can be transformed
- this control is optional, I never do it, but if we are concerned about contamination of reagents with plasmids this could help, so I think we should do it if there is extra insert
- More info on control ligation experiments can be found here
- cut vector withOUT ligase: shows background due to uncut vector
Week 4
September
Week 1
Week 2
Week 3









{kind=link}