 "
"
Team:BGU Israel/Bricks
From 2013.igem.org


Overview
Here is a collection of all the parts we have designed and constructed over the course of our project. Not only did we assemble existing parts in order to create more complex Bio bricks, but we created new parts and improved existing ones. Details of each part can be found in the Registry by a click on the Bio Brick in the following table.
| Part Number | Part Name | Type |
|---|---|---|
| Bba_K1223001 | P.A.S.E1 cassette | Project |
| Bba_K1223002 | P.A.S.E2 cassette | Project |
| Bba_K1223003 | KanR (promoter+CDS) | Coding |
| Bba_K1223004 | Lambda cI CDS | Coding |
| Bba_K1223005 | cI translational unit | Translational Unit |
| Bba_K1223006 | HisTag + stop codon | Tag |
| Bba_K1223007 | cI translational unit with His-tag | Reporter |
| Bba_K1223008 | pUC57-P.A.S.E.2 | Plasmid |
| Bba_K1223009 | pUC57 backbone (REVERSED) | Plasmid Backbone |
| Bba_K1223010 | pUC-57-P.A.S.E.1 | Plasmid |
| Bba_K1223011 | ampR translational unit (ampicilin resistance CDS+promoter) | Translational Unit |
| Bba_K1223012 | pKD46 functional unit | Device |
| Bba_K1223013 | Pyrolysyl-tRNA synthetase CDS | Coding |
| Bba_K1223014 | tRNA-Pyl (pylT) gene from Methanosarcina barkeri str. Fusaro | Coding |
References
[1] M. Pedersen, M. Ligowska, K. Hammer, Characterization of the CI repressor protein encoded by the temperate lactococcal phage, Journal of Bacteriology 29, [2010]
We mixed and matched different parts from the registry with our finishing touch to build our state of the art P.A.S.E machinery.
This part was designed to fulfill the self destruct system of P.A.S.E 1. It contains a toxin system based on phage lysis system of holin (BBa_K112805) and lysozyme (BBa_K112301). Holin protein causes "pores" in the inner membrane, which allows lysozyme to access and break down the peptidoglycan of the cell wall, causing lysis and eventually death. The toxins are regulated by cI regulated promoter (BBa_R0051). This part is designed to integrate into the cell's genome via homologous recombination and therefore it contains homologous regions at its ends. Kanamycin resistance was added for selectivity. Therefore, when transforming in bacteria only the cells that have gone through double recombination with the insert will survive.
 The part was characterized through sequencing and restriction digest with BamHI and EcorRV.
The part was characterized through sequencing and restriction digest with BamHI and EcorRV.

This part was designed to function as a biological timer for our P.A.S.E 1 system. It includes an assembly of two existing parts from the Registry, Lac/Ara-1 IPTG Inducible promoter (BBa_K354000) and LVA-tagged cI repressor protein (BBa_K327018). In order to extend the half life of the cI protein, we removed the LVA tail.
The part was characterized through sequencing and restriction digest with Pvull and HindIII. In addition, the promoter’s performance under the presence of IPTG and Arabinose was analyzed via comassie staining. The cI protein is 29 KDa.

This part is the heart of the P.A.S.E. 2 system. It is intended to be incorporated into E.coli BL21 genome by recombineering, and therefore has a homologous region in each side that direct the recombination into the right place in the genome. This part is used to replace the native promoter and regulatory sequences upstream of the CDS of the TyrS gene that encodes for the enzyme Tyrosine Synthetase. The native promoter is replaced with an IPTG/Arabinose induced promoter (BBa_k354000) and BBa_b0034 RBS. In addition, it has a kanamycin resistance gene (KanR) to aid in selection of the desired transformants.
The sixth amino acid in the TyrS sequence was replaced with an amber stop codon (TAG). The TAG stop codon is used for the incorporation of unnatural amino acids (UAAs) into the protein sequence. This system gives us better control of the translation process and prevents expression of TyrS when the unnatural amino acid is not present in the medium.
 The fail proof aspect of this system is achieved by creating a logic AND gate which is depended on IPTG/Arabinose AND UAA in order to synthesize an active TyrS protein.
The fail proof aspect of this system is achieved by creating a logic AND gate which is depended on IPTG/Arabinose AND UAA in order to synthesize an active TyrS protein.
 The part was characterized through sequencing and restriction digest with BamHI and XbaI.
The part was characterized through sequencing and restriction digest with BamHI and XbaI.

Improving existing parts
Purifying a specific protein from the cell and analyzing its expression requires precise and often expensive tools and ability. Attaching a certain tag to the protein usually simplifies this process and makes it much easier. However, some tags usually have an effect on the function and activity of the protein such as shorting its half life or disrupting its function.
This part is an improvement of the existing BBa_K327018 LVA tagged cI repressor protein. We added a his tag instead of the LVA tag in order to offer a convenient way to study the protein without damaging it. The his tag is located at its C-terminal, therefore having no effect on its function [1]. The part was characterized through comassie staining and western blotting with His probe anti-histag antibody.

 comassie staining western blotting
comassie staining western blotting
Genetic code expansion using stop codon (Amber) suppression in bacteria
Natural translation process is achieved via conservative mRNA-tRNA codon-anticodon specific base pairing; the meaning of each codon is interpreted mainly through stringent substrate specificity of Aminoacyl tRNA synthetases (AARS) in the aminoacylation reaction. This reaction is an interpretation level in the context of the flow of genetic information transmission.[1]
In order to incorporate the #21 man made, synthetic Unnatural amino acid (UAA) one must first find a way to expand the genetic code to add a translational sense codon for that amino acid. We used a method called "Stop codon suppression"[2] developed by Prof. Peter Schultz and co-workers. This method uses orthogonal tRNAcua and Aminoacyl-tRNA synthetase from archaea. Orthogonal means that these components do not interact with the different host organism’s cellular pathways.
Orthogonality:
Since the PylRS originating from archea must be orthogonal in the host cell, our BioBricks could and should be used only in bacteria [2]. There is no orthogonality between archea and mammalian cells for example.
Use in our project:
The following parts were used by us to incorporate the unnatural amino acid proparagyl-L-lysine (as a model UAA) into our PASE 2 essential protein – TyrRS. When one of the following parts or the UAA itself are missing, one of the logical AND gate (Link to PASE2 project description) conditions are not met and essential protein translation is disabled.
Pyrolysyl tRNA synthetase (PylRS) from Methanosarcina barkeri str. Fusaro (Archea):
In well-studied model organisms such as E.coli , yeast and in mammalian cells, the natural AARS catalyse via the aminoacylation reaction the amino acid activation and accurate biosynthesis of aminoacyl-tRNA, the immediate precursors for encoded proteins. Note that each AARS should select its "own" (i.e cognate) amino acid (AA) which is subsequently covalently linked to the cognate tRNA isoacceptor "Fished" from the cellular pool. Immediately after their dissociation from AARS, the aminoacyl tRNA are shuttled or channeled to the ribosome where the anticodon is matched to the mRNA codon and the tRNA is deacylated , with the amino acid being added as the next residue of the a nascent protein chain.[1]
In our project we used a class 2 AARS that is capable of charging Pyrolysine (Pyl – a natural, but rare, amino acid) and several unnatural Amino acids (Fig.1) onto the tRNAcuapyl.
PylRS substrates:
The PylRS can use different derivatives of Pyrolysine with high specificity and fidelity, The substrates of the PylRS are [3]:
 As can be seen in Fig. 2 the aminoacylation of the tRNA by the PylRS is specific and selective to the latter substrates[3].
As can be seen in Fig. 2 the aminoacylation of the tRNA by the PylRS is specific and selective to the latter substrates[3].
 In Fig.2 tRNA were incubated with various lysine derivatives and PylRS, Next all samples were subject to acidic PAGE and stained with methylene blue. All samples that have 2 bends have been aminoacylated with the relevant lysine derivative.
In our study proparagyl-lysine (figure 1, (8)) was used as a model amino acid.
PylRS Structure:
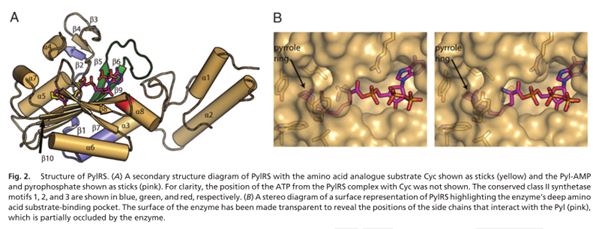
The crystal structure of the entire protein still eludes structural biologists but the crystal structure of its catalytic domain has been recently solved (Fig.3). The PylRS catalytic domain structure analysis reveals that it utilizes a deep hydrophobic pocket for recognition of the Pyl side chain (The pocket binds both tRNA and the amino acid and catalyzes the aminoacylation reaction between them) [4].
In Fig.2 tRNA were incubated with various lysine derivatives and PylRS, Next all samples were subject to acidic PAGE and stained with methylene blue. All samples that have 2 bends have been aminoacylated with the relevant lysine derivative.
In our study proparagyl-lysine (figure 1, (8)) was used as a model amino acid.
PylRS Structure:
The crystal structure of the entire protein still eludes structural biologists but the crystal structure of its catalytic domain has been recently solved (Fig.3). The PylRS catalytic domain structure analysis reveals that it utilizes a deep hydrophobic pocket for recognition of the Pyl side chain (The pocket binds both tRNA and the amino acid and catalyzes the aminoacylation reaction between them) [4].
 This Biobrick is the coding sequence for the Pyrolysyl tRNA synthetase enzyme from the archaea Methanosarcina barkeri str. fusaro. As mentioned earlier, the enzyme selectively 'loads' the amino acid, pyrolysine (and other synthetic substrates) onto its cognate tRNA for subsequent incorporation into proteins during the translation process in the ribosome. The tRNA synthetase is part of the unnatural amino acid (UAA) incorporation machinery along its cognate tRNA molecule (BBa_K1223014).
This Biobrick is the coding sequence for the Pyrolysyl tRNA synthetase enzyme from the archaea Methanosarcina barkeri str. fusaro. As mentioned earlier, the enzyme selectively 'loads' the amino acid, pyrolysine (and other synthetic substrates) onto its cognate tRNA for subsequent incorporation into proteins during the translation process in the ribosome. The tRNA synthetase is part of the unnatural amino acid (UAA) incorporation machinery along its cognate tRNA molecule (BBa_K1223014).
BBa_K1223014 – tRNAcua pyl (pylT) gene from Methanosarcina barkeri:
This biobrick is the DNA coding sequence for the tRNAcua pyl of Pyrolysine from the Archaea Methanosarcina barkeri str fusaro. This tRNA is a part of the machinery that is used to incorporate pyrolysine (and other substrates) into proteins in bacteria. The PylRS that charges the tRNA with pyrrolysine can be found in biobrick BBa_k1223013. The tRNA naturally contains the anticodon CUA (Fig.4) – Thus it recognizes TAG as its sense codon when incorporating the UAA. With the introduction of this tRNAcua pyl from archaea into bacteria it can compete with the bacterial release factors in the recognition of the UAG codon of the mRNA. This competition can manipulate the bacterial translational machinery (stop codon suppression) and thus render the TAG non-sense codon (Amber stop codon) to a Sense codon for UAA incorporation. UAA incorporation efficiency: For efficient stop codon suppression the expression of tRNA¬cua¬pyl levels should be high. For this reason the pylT gene must be cloned (along with the PylRS) into a high copy number expression plasmid. In addition, multiple copies (6-9) of the gene encoding for the amber suppressor (tRNAcua pyl) can be added to the same backbone.[6]
For the characterization of those parts we designed and carried out two different experiments:
1. To determine and characterize our ability to incorporate an unnatural amino acid (Progargyl lysine) into a model protein site specifically we first used standard mutagenesis methods to add TAG stop codons to various sites in our model gene.
Next we transformed the bacterium with our model protein on an expression plasmid and then transformed the same bacteria with an expression plasmid containing our PylRS and tRNAcua. We have grown bacteria in LB broth overnight with 1mM of expression inducers (IPTG) and 1mM of Propargyl lysine (UAA). Upon incubation we lysed the bacteria and performed a click reaction with a fluorescently labeled azide(See protocols - link) to determine the presence of propargyl lysine in place. In order to visualize the protein, we ran the lysates in an SDS fluorescent PAGE.[7]

- 1st experiment:

Fig. 5. Fluorescent western blot image, using anti-6xHis antibody to visulaize different CueO mutants expressed in the presence (+) and in the absence (-) of UAA. Commassie staining 
Fig. 6. Commasie stained gel image of the same samples shown on the western blot in figure 5. In this experiment we produced both fluorescent western blot and coomassie stained gel (of the same experiment) that shows the specificity of the unnatural amino acid incorporation machinery in four different mutants in the E.coli copper oxidase (CueO). For each mutant we induced the production of the protein (~57kDa) with (+) or without (-) the unnatural amino acid present in the growth medium. It can be seen very clearly from the fluorescent blot that the protein is present only in the culture that were incubated with the unnatural amino acid - except for G450 which appears to be a non-permisive site where there is no incorporation of UUA at all. - 2nd experiment:
 In this Anti-his-tag western blotting we were able to show that there in minimal non-
specific incorporation when there were no UAA. In other words, our logic AND gate 2nd
condition is robust. When our bacteria are in nature they could not translate their
essential protein, but its inactive truncated form.
In this Anti-his-tag western blotting we were able to show that there in minimal non-
specific incorporation when there were no UAA. In other words, our logic AND gate 2nd
condition is robust. When our bacteria are in nature they could not translate their
essential protein, but its inactive truncated form.
Potential applications of Genetic code expansion and UAA:
By the use of site specific incorporation of many different Unnatural amino acids into proteins numerous possibilities open up for synthetic biology and many other fields. Among those possibilities are:
- Probes of Protein Structure and Function: Many biophysical and mechanistic studies require significant quantities of proteins with a probe incorporated at a unique site in a protein. UAA mutagenesis methodology is well suited to many such problems.[8]
- Therapeutic proteins UAA mutagenesis is beginning to find many applications in the generation of therapeutic proteins, where the production of large quantities of homogenously modified protein is desired.[8]
- Protein Evolution with an Expanded Genetic Code It is quite possible that the ability to encode additional amino acids with novel properties would be evolutionarily advantageous, especially since nature’s choice of 20 could have been arbitrarily fixed at the point of transition between communal and Darwinian evolution paradigms and subsequently sustained by the code’s inertia. Furthermore, in the limited scope of laboratory-directed evolution, which concerns only one or few specific functions over a short time rather than general organismal fitness over thousands or millions of years, one can easily envision a selective advantage conferred by additional amino acids. Because the templated assembly of polypeptides from mRNA on the ribosome establishes a direct link between genes (information) and proteins (phenotype), UAA mutagenesis methodology can easily be adapted to the evolution of proteins with novel or enhanced function.[8]
Conclusion
We hope that by characterizing and adding these important natural parts to the iGEM registry we will open new options for iGEM teams in particular and the SynBio community in general.
Additional natural parts
We added two additional new biobricks: Assembling parts to create a new DNA strand always requires taking into account all the restriction sites that exists on each part. Not always is it possible to assemble specific parts for they may contain overlapping restriction sites which do not enable to "cut and paste" between them. In order to overcome this obstacle, there is a need for expanding the selection of options for each part. We contributed to this issue by adding two new parts, though with similar activity to existing ones but with a different sequence. Both parts are a common antibiotic resistance: 1. BBa_K1223003 - kanamycin resistance gene (promoter + CDS) 2. BBa_K1223011 - ampR translational unit (ampicillin resistance CDS + promoter)
References
[1] N. Budisa, “Prolegomena to future experimental efforts on genetic code engineering by expanding its amino acid repertoire.,” Angew. Chem. Int. Ed. Engl., vol. 43, no. 47, pp. 6426–63, Dec. 2004. [2] L. Wang, J. Xie, and P. G. Schultz, “Expanding the genetic code.,” Annu. Rev. Biophys. Biomol. Struct., vol. 35, pp. 225–49, Jan. 2006. [3] O. Nureki, Y. Nakahara, K. Nozawa, H. Hojo, and H. Katayama, “Pyrrolysine Analogs as Substrates for Bacterial Pyrrolysyl-tRNA Synthetase in Vitro and in Vivo,” Biosci. Biotechnol. Biochem., vol. 76, no. 1, pp. 205–208, 2012. [4] J. M. Kavran, S. Gundllapalli, P. O. Donoghue, M. Englert, D. So, and T. A. Steitz, “Structure of pyrrolysyl-tRNA synthetase , an archaeal,” 2007. [5] A. Théobald-Dietrich, M. Frugier, R. Giegé, and J. Rudinger-Thirion, “Atypical archaeal tRNA pyrrolysine transcript behaves towards EF-Tu as a typical elongator tRNA.,” Nucleic Acids Res., vol. 32, no. 3, pp. 1091–6, Jan. 2004. [6] Y. Ryu and P. G. Schultz, “Efficient incorporation of unnatural amino acids into proteins in Escherichia coli,” vol. 3, no. 4, pp. 263–266, 2006. [7] D. P. Nguyen, H. Lusic, H. Neumann, P. B. Kapadnis, A. Deiters, and J. W. Chin, “Genetic encoding and labeling of aliphatic azides and alkynes in recombinant proteins via a pyrrolysyl-tRNA Synthetase/tRNA(CUA) pair and click chemistry.,” J. Am. Chem. Soc., vol. 131, no. 25, pp. 8720–1, Jul. 2009. [8] C. C. Liu and P. G. Schultz, “Adding new chemistries to the genetic code.,” Annu. Rev. Biochem., vol. 79, pp. 413–44, Jan. 2010.
We are currently working on inserting the Lac/Ara-1 IPTG inducible promoter (BBa_K354000) into a pGFPuv plasmid through side-directed mutagenesis. This is done to improve the BBa_K354000 BioBrick by attaching to it a GFP reporter gene and by improving our ability to characterize it. After a few unsuccessful tries, iGEM team Paris_Bettencourt kindly offered us their help. They are currently helping us characterizing this part by putting a GFP gene under the expression of this promoter. Additionally, we intend to characterize and develop the Lambda red incorporation machinery (pKD Bba_K1223012) used in our project to a working and well defined biobrick .
Continue the journey: read about our achievements.
