Results
Team Parts Sandbox
| Name |
Type |
Description |
Designer |
Length |
Favorite Part |
| [http://parts.igem.org/Part:BBa_K1210000 BBa_K1210000] |
Device |
LacI-CFP-PK401-YFP |
Zak Stinson |
1955 |
Yes |
| [http://parts.igem.org/Part:BBa_K1210001 BBa_K1210001] |
RNA Part |
PK401 Pseudoknot |
Zak Stinson |
93 |
|
| [http://parts.igem.org/Part:BBa_K1210002 BBa_K1210002] |
Device |
Enhanced Lumazine Synthase (ELS) Expression Construct |
Harland Brandon |
828 |
|
Expression and Characterization of PK401 (BBa_K1210000)
PK401 Overexpression
The PK401 construct was overexpressed to test and characterize the construct for frameshifting ability. To do this, cultures of E. coli DH5α containing either the PK401 plasmid or an empty control plasmid were grown from glycerol stocks overnight at 37°C in 50 mL LB media containing Kanamycin (100 µg/mL). These cultures were used the next day to inoculate 500 mL of LB media to a starting optical density at 600 nm (OD600) near 0.05. The OD600 was monitored and induced with 1 mM IPTG once reaching an OD600 of 0.6 (Fig. 1). Growth was monitored hourly, and equivalent amounts of cells were taken as samples for SDS-PAGE analysis up to 5 h after induction. The cultures were harvested by centrifuging at 5000 x g and shock frozen to store at -80°C.
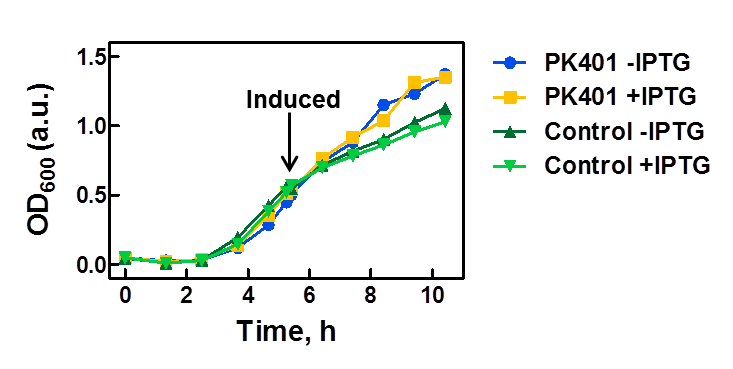
Figure 1. Growth curve of PK401 construct. E. coli DH5α cells containing the PK401 plasmid or a control plasmid were grown at 37°C in LB media. The OD600 was monitored and the cultures were induced with 1 mM IPTG when the OD600 had reached 0.6. The cultures were grown for an additional 5 h, and a sample of 1 OD600 equivalent of cells were taken at each hour after induction.
The samples taken for SDS-PAGE analysis were pelleted and resuspended in 80 µL 0.1 M Tris-HCl pH 8.5 containing 5 M urea and 20 µL SDS-PAGE gel-loading buffer. The samples were then analyzed on a 12% SDS-PAGE and stained with Coomassie blue to confirm overexpression of the non-frameshifted and -1 frameshifted protein products from the PK401 construct (Fig. 2). The non-frameshifted product (CFP) has an expected size of 29 kD, and the -1 frameshifted product (a fusion protein of CFP-PK401-YFP) has an expected size of 60 kD. Bands of increasing intensity after induction with IPTG were seen at approximately 30 kD and 60 kD, corresponding to both the non-frameshifted and -1 frameshifted product. These same bands were seen in the uninduced samples, however this could be due to the expression being controlled by the pLacI promoter, which is known to give leaky expression.
To estimate the frameshifting frequency of PK401, the relative band intensities of the 60 kD protein were compared to that of the 29 kD protein. This resulted in a calculated frameshift efficiency of 21 ± 5%, indicating that 21% of the translated product was frameshifted into the -1 frame to produce the CFP-PK401-YFP fusion protein.
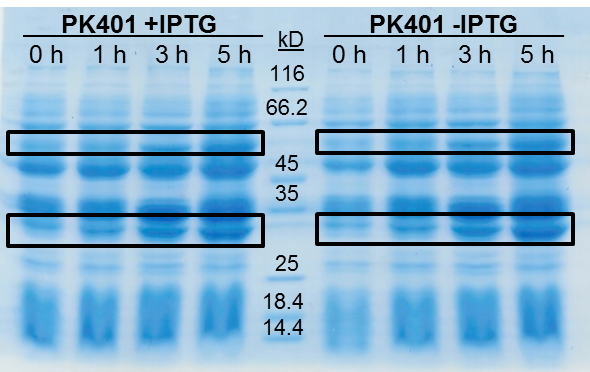
Figure 2. Over-expression of non-frameshifted and -1 frameshifted protein products from the PK401 construct. Equivalent amounts of cells at 0, 1, 3, and 5 h after induction with IPTG (PK401 +IPTG) were analyzed by 12% SDS-PAGE. The same time samples from the uninduced culture were also analyzed (PK401 –IPTG). Black boxes indicate bands of increasing intensity that migrated with an approximate molecular weight of 60 kD and 30 kD, corresponding to the -1 frameshifted CFP-PK401-YFP fusion product and the non-frameshifted CFP product, respectively.
CFP and YFP Fluorescence from PK401 Overexpression
To confirm that the protein products seen in the overexpression of the PK401 construct corresponded to the expected fluorescent proteins, fluorescence spectra of the cell lysates from overexpression were measured. The cells from the overexpression were lysed using 1 mg/mL lysozyme and 12.5 mg/g sodium deoxycholate. DNase was added to degrade the DNA followed by centrifugation at 3000 x g for 30 min. The supernatant was collected and centrifuged further at 30 000 x g for 45 min. This supernatant was then used for fluorescence measurements.
The supernatants were diluted 100-fold in the buffer used for cell opening. Excitation and emission scans were measured for all samples in the CFP and YFP range in order to confirm the presence of both fluorescent proteins. CFP was excited at 430 nm, and emission was monitored from 445-650 nm (Fig. 3). YFP was excited at 510 nm, and emission was monitored from 525-650 nm (Fig. 4). These spectra were measured for the cell opening buffer and for the induced and uninduced samples of the cells that contained the PK401 construct, as well as the cells containing the control plasmid.
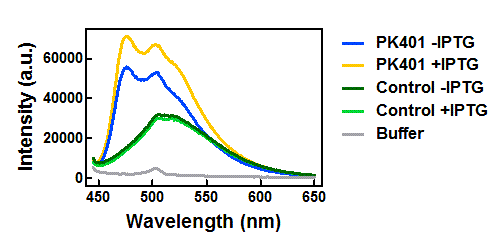
Figure 3. Emission spectra after excitation at 430 nm. Cell lysates from the cultures used for overexpression were excited near the excitation maximum of CFP (at 430 nm) and the emission was monitored from 445-650 nm. The spectra shown include the uninduced PK401 construct (blue), induced PK401 construct (yellow), uninduced control (dark green), induced control (lightgreen), and cell opening buffer (grey).
The cell lysates from the PK401 overexpression show the characteristic emission spectrum of CFP upon excitation at 430 nm. There is an additional shoulder in the spectrum near 527 nm, which is the emission maximum of YFP. When YFP was excited directly in the PK401 overexpression samples, the characteristic emission spectrum of YFP was seen, indicating that both CFP and YFP were expressed in these samples. The fluorescence from YFP could only result from a frameshift during expression of the PK401 transcript, indicating that the pseudoknot worked as expected to induce a -1 frameshift during translation.
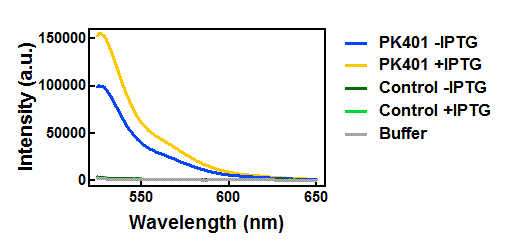
Figure 4. Emission spectra after excitation at 510 nm. Cell lysates from the cultures used for overexpression were excited near the excitation maximum of YFP (at 510 nm) and the emission was monitored from 525-650 nm. The spectra shown include the uninduced PK401 construct (blue), induced PK401 construct (yellow), uninduced control (dark green), induced control (lightgreen), and cell opening buffer (grey). The characteristic emission maximum of YFP is at 527 nm, which was observed in the spectra from the PK401 samples.
Upon excitation of the cell lysates from the control plasmid cultures at 430 nm, a fluorescence signal of half the intensity of the PK401 cell lysates was observed with a plateau from approximately 500-520 nm. This did not correspond to the emission maximum of CFP or YFP, so the signal was likely due to other cellular components. Additionally, when the sample was excited at 510 nm, there was no detectable emission signal, indicating that YFP was not present in these samples.
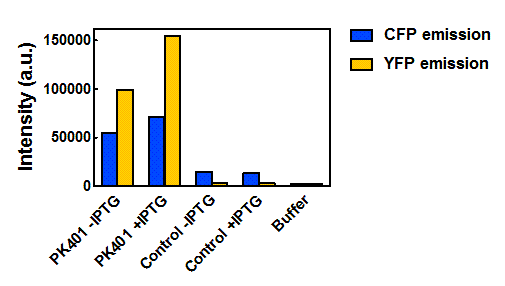
Figure 5. Maximum fluorescence intensity of overexpression cell lysates. Intensities of cell lysates after excitation at 430 nm and monitoring at 475 nm (CFP fluorescence, blue bars) and excitation at 510 nm and emission at 527 nm (YFP fluorescence, yellow bars).
When the peak intensities from each sample were analyzed individually (Fig. 5), more prominent fluorescence measurements were observed for the PK401 cell lysates than for the control cell lysates. Excitation at 430 nm resulted in a fluorescence emission at 475 nm, which is a characteristic emission maximum of CFP, for the PK401 samples, but not for the control samples. Additionally, direct excitation and emission from YFP was seen for the PK401 cell lysates, but no fluorescence emission was seen for the control samples after excitation at 510 nm. This further indicates that the PK401 pseudoknot induced a -1 frameshift during translation of the PK401 transcript to produce both CFP and YFP.
Generating a Pseudoknot Library
Generating a Library of Pseudoknots with Variable Frameshifting Frequencies
To make better use of the frameshifting capability of the pseudoknot, we are also generating a randomized library of pseudoknot sequences that can be characterized for frameshifting frequency. It is our goal to create a library of pseudoknots that have a range of frameshifting frequencies to better facilitate broad application of this part. To do this, the PK401 sequence will be mutagenized using error-prone PCR to generate a library of primers that will be used to amplify the plasmid containing the original sequence. The newly generated plasmids contained mutated PK401 sequences will be transformed into E. coli cells, sequenced, and characterized for their specific frameshifting efficiency using a construct such as BBa_K1210000.
Optimizing Conditions for the Error-Prone PCR
To generate a library with high variability of sequence, conditions for the error-prone PCR had to first be optimized. We wanted to disrupt the PCR experiment enough to produce a large variety of primer sequences, but not too much so that the PCR was no longer successful. To do this, we used a temperature gradient to determine the annealing temperature that would best facilitate the amplification of the expected product. Figure 6 shows the resulting PCR products from the temperature gradient reactions, using temperatures from 41.9-62.1°C. The expected size of the PCR product is 93 bp, which was seen in the first three reaction samples. Various unspecific PCR products were seen in the remaining reactions samples. For this reason, the temperature chosen for the subsequent error-prone PCRs was 43.6°C, the temperature used to produce the PCR product in lane 3.
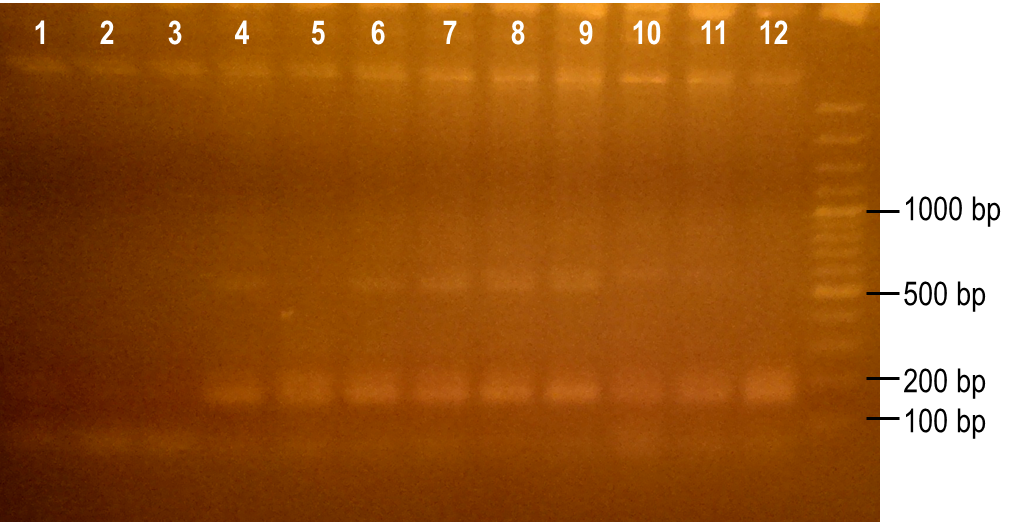
Figure 6. Determining the ideal temperature for error-prone PCR reactions. PCR reactions were performed using various annealing temperatures from 41.9-62.1°C (lanes 1-12). The primers used in the reaction flank the PK401 pseudoknot, making the expected product size 93 bp. The temperature from the reaction in lane 3 (43.6°C) was selected for subsequent error-prone PCRs.
Next, we modulated a few of the typical PCR conditions to induce errors during the reaction. First, we used Taq polymerase, which is known to have higher error rates than higher fidelity polymerases commonly used in PCRs. Second, we added MnCl2 to the reaction to further hinder the accuracy of the polymerase. Finally, different ratios of deoxynucleotides were used in the reactions, either by increasing the dGTP concentration or by using a higher concentration of both dTTP and dCTP. A number of these conditions worked to produce the expected PCR product at 93 bp (Fig. 7, lanes 1-4, 6). The conditions tested in lanes 1-4 included MnCl2 at concentrations of 0-0.45 mM, and lane 6 tested the effect of increasing dGTP concentrations to 1 mM while the other dNTPs were at a concentration of 0.2 mM.
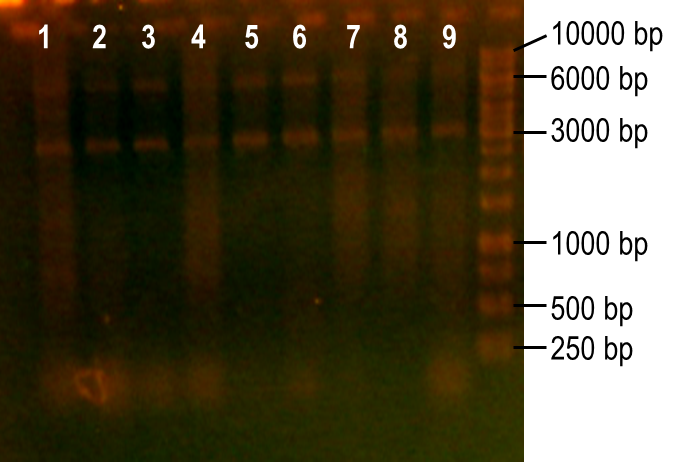
Figure 7. Error-prone PCR of PK401 sequence. PCR conditions were adjusted to induce errors in the PK401 sequence. Reactions were performed with the following modified condtions: lanes 1-6 used MnCl2 concentrations from 0-0.65 mM, lanes 4-6 used dGTP concentrations that were increased to 0.7-1.2 mM, lane 7 used increased concentrations of dTTP and dCTP, lane 8 used increased concentrations of dGTP only, and lane 9 used 0.65 mM MnCl2 only.
Now that we have determined the conditions for the error-prone PCR, we will perform a large-scale amplification so that we can purify the resulting PK401 primers by extracting it from an agarose gel. The primers will then be used in a high fidelity PCR reaction to amplify the entire plasmid, which will create the library of PK401 sequences. The final steps will be to transform the final PCR products into E. coli cells, screen them for frameshifting frequency using a similar construct as BBa_K1210000, and sequence the pseudoknot before submitting the parts to the registry to complete the library of frameshifting elements.
 "
"







