 "
"
Team:SDU-Denmark/Tour51
From 2013.igem.org
| Line 43: | Line 43: | ||
<div class="galleryMedium alignCenter"> | <div class="galleryMedium alignCenter"> | ||
| - | <a class="galleryImg" target="_blank | + | <a class="galleryImg" target="_blank" |
href="https://static.igem.org/mediawiki/2013/f/f6/SDU2013_Cloning_2.png" title="Figure 2 - USER cloning was used to produce the construct pSB1C3-Plac-dxs (B. subtilis). Our tests of the construct showed unfortunately two restriction sites within the coding sequence."> | href="https://static.igem.org/mediawiki/2013/f/f6/SDU2013_Cloning_2.png" title="Figure 2 - USER cloning was used to produce the construct pSB1C3-Plac-dxs (B. subtilis). Our tests of the construct showed unfortunately two restriction sites within the coding sequence."> | ||
<img src="https://static.igem.org/mediawiki/2013/f/f6/SDU2013_Cloning_2.png"> | <img src="https://static.igem.org/mediawiki/2013/f/f6/SDU2013_Cloning_2.png"> | ||
| + | Fig. 2 | ||
</a> | </a> | ||
| - | <a class="galleryImg" target="_blank | + | <a class="galleryImg" target="_blank" |
href="https://static.igem.org/mediawiki/2013/c/ce/SDU2013_Cloning_3.png" title="Figure 3 - Site directed mutagenesis was performed to remove the restriction sites, which ensured that the gene complied with iGEM standards. The gel picture shows the expected length around 2000 bp."> | href="https://static.igem.org/mediawiki/2013/c/ce/SDU2013_Cloning_3.png" title="Figure 3 - Site directed mutagenesis was performed to remove the restriction sites, which ensured that the gene complied with iGEM standards. The gel picture shows the expected length around 2000 bp."> | ||
<img src="https://static.igem.org/mediawiki/2013/c/ce/SDU2013_Cloning_3.png"> | <img src="https://static.igem.org/mediawiki/2013/c/ce/SDU2013_Cloning_3.png"> | ||
| + | Fig. 3 | ||
</a> | </a> | ||
| - | <a class="galleryImg" target="_blank | + | <a class="galleryImg" target="_blank" |
href="https://static.igem.org/mediawiki/2013/b/bc/SDU2013_Cloning_4.png" title="Figure 4 - The gel-picture shows the result for the test digestion. Linearized plasmid has the expected length around 5500 bp, and the EcoRI and PstI cut plasmid has expected lengths around 2000 bp and 3500 bp."> | href="https://static.igem.org/mediawiki/2013/b/bc/SDU2013_Cloning_4.png" title="Figure 4 - The gel-picture shows the result for the test digestion. Linearized plasmid has the expected length around 5500 bp, and the EcoRI and PstI cut plasmid has expected lengths around 2000 bp and 3500 bp."> | ||
<img src="https://static.igem.org/mediawiki/2013/b/bc/SDU2013_Cloning_4.png"> | <img src="https://static.igem.org/mediawiki/2013/b/bc/SDU2013_Cloning_4.png"> | ||
| + | Fig. 4 | ||
</a> | </a> | ||
| - | + | Figures 8 - 10. | |
| - | Figures | + | |
</div> | </div> | ||
| + | |||
| + | |||
| + | |||
</p> | </p> | ||
<p> | <p> | ||
Revision as of 01:24, 4 October 2013
Cloning
The beauty of creation
To clarify the progression of our cloning, the results will not be presented chronologically, but rather in a logical manner, starting with the simplest constructs. This is not an exhaustive list, but merely a presentation to clarify our work and give insight into our thoughts. Please consult our submitted parts-page (dig deeper), as well as our comprehensive protocols to gain full insight into our work. We have done our utmost to edit the gel photos in a responsible manner. Should questions arise, originals can be found in our protocols.
 Figure 1.
Figure 1.
pSB1C3-Plac-dxs (E. coli)
The dxs from E. coli was already present in parts registry (BBa_K118000), which simplified our aim to increase expression of the gene. However, the middle part of the brick was unsequenced, so this was an obvious focal point. We successfully completed the sequencing and the full sequence has been entered into part registry. User cloning was successfully used to clone PLac together with E.coli dxs. The picture shows the colony PCR result for the cloning and subsequent transformation. The gel picture shows the expected length around 2300 bp including verification primers (Fig. 1).
pSB1C3-Plac-dxs (B. subtilis)
The literature suggested that dxs from B. subtilis performed better than its E. coli counterpart. Therefore, a colony PCR was performed on the B. subtilis strain 168. USER cloning was used to produce the construct pSB1C3-Plac-dxs (B. subtilis). Our tests of the construct showed unfortunately two restriction sites within the coding sequence (Fig. 2).Site directed mutagenesis was performed to remove the restriction sites, which ensured that the gene complied with iGEM standards. The gel picture shows the expected length around 2000 bp (Fig. 3).
Then, our focus moved to expression control: Subsequent testing proved that the inherent amount of LacI (the repressor of the lactose promoter) in MG1655 was insufficient in the presence of our construct in a high-copy plasmid (see characterization). Therefore, LacI with an LVA tag was taken from parts registry (BBa_C0012) and cloned into our construct with its own promoter and terminator. pSB1C3-Pcon-LacI:LVA-term-Plac-dxs(sub) was born. The gel-picture shows the result for the test digestion. Linearized plasmid has the expected length around 5500 bp, and the EcoRI and PstI cut plasmid has expected lengths around 2000 bp and 3500 bp (Fig. 4).
 Fig. 2
Fig. 2
 Fig. 3
Fig. 3
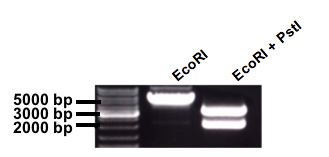 Fig. 4
Fig. 4
This brick would allow us to overcome the first rate-limiting step of the MEP pathway in a regulatable manner.
 Figure 5.
Additionally, we observed indications that LacI:LVA was an inferior repressor to MG1655’s own LacI. As a consequence, we performed yet another colony PCR to amplify natural LacI, and clone this into our construct in place of LacI:LVA. Lane 7 of the gel picture shows colony PCR result for the transformation with pSB1C3-Pcon-LacI(N)-term-Plac-dxs(B.sub). A band appeared just above 3000 bp as expected for this construct (Fig. 5).
Figure 5.
Additionally, we observed indications that LacI:LVA was an inferior repressor to MG1655’s own LacI. As a consequence, we performed yet another colony PCR to amplify natural LacI, and clone this into our construct in place of LacI:LVA. Lane 7 of the gel picture shows colony PCR result for the transformation with pSB1C3-Pcon-LacI(N)-term-Plac-dxs(B.sub). A band appeared just above 3000 bp as expected for this construct (Fig. 5).
As a final test of this hypothesis, the plasmid was transformed into KG22, an E. coli strain with natural abundance of LacI. This is further discussed in our characterization of LacI on the next page in the tour.
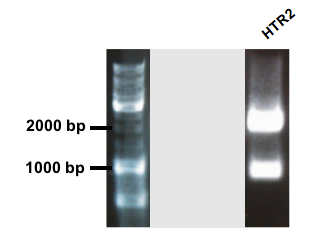 Figure 6.
Figure 6.
pSB1C3-Para-HRT2
Genestript performed the synthesis of HRT2 polyprenyltranferase with a flagtag added in the end of the sequence, an RBS and an arabinose promoter in front. Unfortunately, the stop codon of the gene was not removed, leaving the tag unfunctionable. The coding sequence of HRT2 was amplified with primers adding prefix and suffix to the sequence. The gel picture shows two bands. The band just above 2000 bp corresponds to the expected length of shipped pUC57 plasmid and the second band around 900 bp is the expected length for the coding HTR2 sequence (Fig. 6).
 Figure 7.
In order to assay optimized control of the expression from the arabinose promoter and thus HTR2, we build and added an AraC device. A colony PCR was performed on MG1655 to obtain AraC, which was cloned into the construct with its own promoter and terminator. The construct design was: psB1C3-Pcon-AraC-term-Para-HRT2-FT. The colony PCR for the transformation with the construct shows a band just above 2000 as expected (Fig. 7).
Figure 7.
In order to assay optimized control of the expression from the arabinose promoter and thus HTR2, we build and added an AraC device. A colony PCR was performed on MG1655 to obtain AraC, which was cloned into the construct with its own promoter and terminator. The construct design was: psB1C3-Pcon-AraC-term-Para-HRT2-FT. The colony PCR for the transformation with the construct shows a band just above 2000 as expected (Fig. 7).
We had great difficulty cloning the construct into pSB1A3, and therefore chose pSB1K3 as our plasmid for the production system.
Reporter systems
To check the expression profile of our constructs, we used two distinct methods: 1) For the dxs construct, protein fusion with a reporter gene, coupled with microscopy or FACS was used, and 2) for the HRT2 construct, Northern blots were performed. To see the results of these tests, see Characterization.For our reporter system, we produced pSB1C3-Pcon-dxs (B. subtilis)-AmilCP. The AmilCP, however, proved to be a very poor fusion reporter gene, and we abandoned this construct to focus our attention on GFP. A number of constructs were successfully made, corresponding to the evolution of our design outlined above (Fig. 8-10).
Figure 8 - Colony PCR result for pSB1C3-plac-dxs(b.sub)-linker-GFP
Figure 9 - Test digest of pSB1C3-LacI:LVA-plac-dxs(b.sub)-linker-GFP
Figure 10 - Test digest of pSB1C3-LacI(N)-plac-dxs(b.sub)-linker-GFP
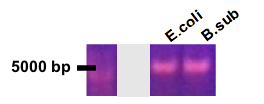 Fig. 8
Fig. 8
 Fig. 9
Fig. 9
 Fig. 10
Fig. 10
A list of our primers