"
"
Team:TU Darmstadt/modelling/Structure
From 2013.igem.org
| Line 376: | Line 376: | ||
<body> | <body> | ||
| - | <font size="5" color="#F0F8FF" face="Arial regular">LssmOrange</font> | + | <font size="5" color="#F0F8FF" face="Arial regular">LssmOrange - first modelling</font> |
</body> | </body> | ||
| Line 403: | Line 403: | ||
<!-- Tabelle --> | <!-- Tabelle --> | ||
| - | + | ||
</body> | </body> | ||
| Line 446: | Line 446: | ||
</table> | </table> | ||
</body> | </body> | ||
| + | |||
| + | |||
| + | |||
| + | <!-- Tabelle --><br> | ||
| + | <br> | ||
| + | |||
| + | <body> | ||
| + | <html> | ||
| + | <table border="1"> | ||
| + | <tr> | ||
| + | <th><b>Check Type</b></th> | ||
| + | <td><b>Quality Z-score</b></td> | ||
| + | <td><b>Comment</b></td> | ||
| + | |||
| + | </tr> | ||
| + | <tr> | ||
| + | <th>Dihedrals</th> | ||
| + | <th>WERT</th> | ||
| + | <td>BLA</td> | ||
| + | |||
| + | </tr> | ||
| + | <tr> | ||
| + | |||
| + | <th>Packing 1D</th> | ||
| + | <th>WERT</th> | ||
| + | <td>BLA</td> | ||
| + | </tr> | ||
| + | <tr> | ||
| + | |||
| + | <th>Packing 3D</th> | ||
| + | <th>WERT</th> | ||
| + | <td>BLA</td> | ||
| + | </tr> | ||
| + | |||
| + | <tr> | ||
| + | |||
| + | <th>Overall</th> | ||
| + | <th>WERT</th> | ||
| + | <td>BLA</td> | ||
| + | </tr> | ||
| + | |||
| + | </table> | ||
| + | </body> | ||
| + | |||
| + | |||
| + | |||
| + | <!-- Tabelle --><br> | ||
| + | <br> | ||
| + | |||
| + | <body> | ||
| + | <html> | ||
| + | <table border="1"> | ||
| + | <tr> | ||
| + | <th><b>Check Type</b></th> | ||
| + | <td><b>Quality Z-score</b></td> | ||
| + | <td><b>Comment</b></td> | ||
| + | |||
| + | </tr> | ||
| + | <tr> | ||
| + | <th>Dihedrals</th> | ||
| + | <th>WERT</th> | ||
| + | <td>BLA</td> | ||
| + | |||
| + | </tr> | ||
| + | <tr> | ||
| + | |||
| + | <th>Packing 1D</th> | ||
| + | <th>WERT</th> | ||
| + | <td>BLA</td> | ||
| + | </tr> | ||
| + | <tr> | ||
| + | |||
| + | <th>Packing 3D</th> | ||
| + | <th>WERT</th> | ||
| + | <td>BLA</td> | ||
| + | </tr> | ||
| + | |||
| + | <tr> | ||
| + | |||
| + | <th>Overall</th> | ||
| + | <th>WERT</th> | ||
| + | <td>BLA</td> | ||
| + | </tr> | ||
| + | |||
| + | </table> | ||
| + | </body> | ||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
Revision as of 16:13, 4 October 2013
Homology Modelling
While our proteins are functionally described in literature and during the IGEM competition, only part of the structures are available in the protein data bank. For further work and visualizations, protein structures are indispensable. We used Yasara Structure [1] to calculate 3-dimensional structures of all of our proteins for the IGEM.
Workflow
Description how our Yasara script calculates homology model[7]:

- Sequence is PSI-BLASTed against Uniprot [2]
- Calculation of a position-specific scoring matrix (PSSM) from related sequences
- Using the PSSM to search the PDB for potential modeling templates
- The Templates are ranked based on the alignment score and the structural quality[3]
- Deriving additional information’s for template and target (prediction of secondary structure, structure-based alignment correction by using SSALN scoring matrices [4]).
- A graph of the side-chain rotamer network is built, dead-end elimination is used to find an initial rotamer solution in the context of a simple repulsive energy function [5]
- The loop-network is optimized using a high amount of different orientations
- Side-chain rotamers are fine-tuned considering electrostatic and knowledge-based packing interactions as well as solvation effects.
- An unrestrained high-resolution refinement with explicit solvent molecules is run, using the latest knowledge-based force fields[6].
Application
All these steps are performed to every template used for the modeling approach. For our project we set the maximum amount of templates to 20. Every derived structure is evaluated using an average per-residue quality Z-scores. At last a hybrid model is built containing the best regions of all predictions. This procedure make prediction’s accurate and thus more realistic. For the evaluation we used the Yasara Z-scores.A Z-score describes how many standard deviations the model quality is away from the average high-resolution X-ray structure. Negative values indicate that the homology model looks worse than a high-resolution X-ray structure. The overall Z-scores for all models have been calculated as the weighted averages of the individual Z-scores using the formula Overall = 0.145*Dihedrals + 0.390*Packing1D + 0.465*Packing3D [7].
Parameters
We used the Yasara script hm_build.mcr for the model creation with the following parameters:
- Modeling speed (slow = best): Slow
- Number of PSI-BLAST iterations in template search (PsiBLASTs): 3
- Maximum allowed PSI-BLAST E-value to consider template (EValue Max): 0.5
- Maximum number of templates to be used (Templates Total): 20
- Maximum number of templates with same sequence (Templates SameSeq): 1
- Maximum oligomerization state (OligoState): 4 (tetrameric)
- Maximum number of alignment variations per template: (Alignments): 5
- Maximum number of conformations tried per loop (LoopSamples): 50
- Maximum number of residues added to the termini (TermExtension): 10
Results
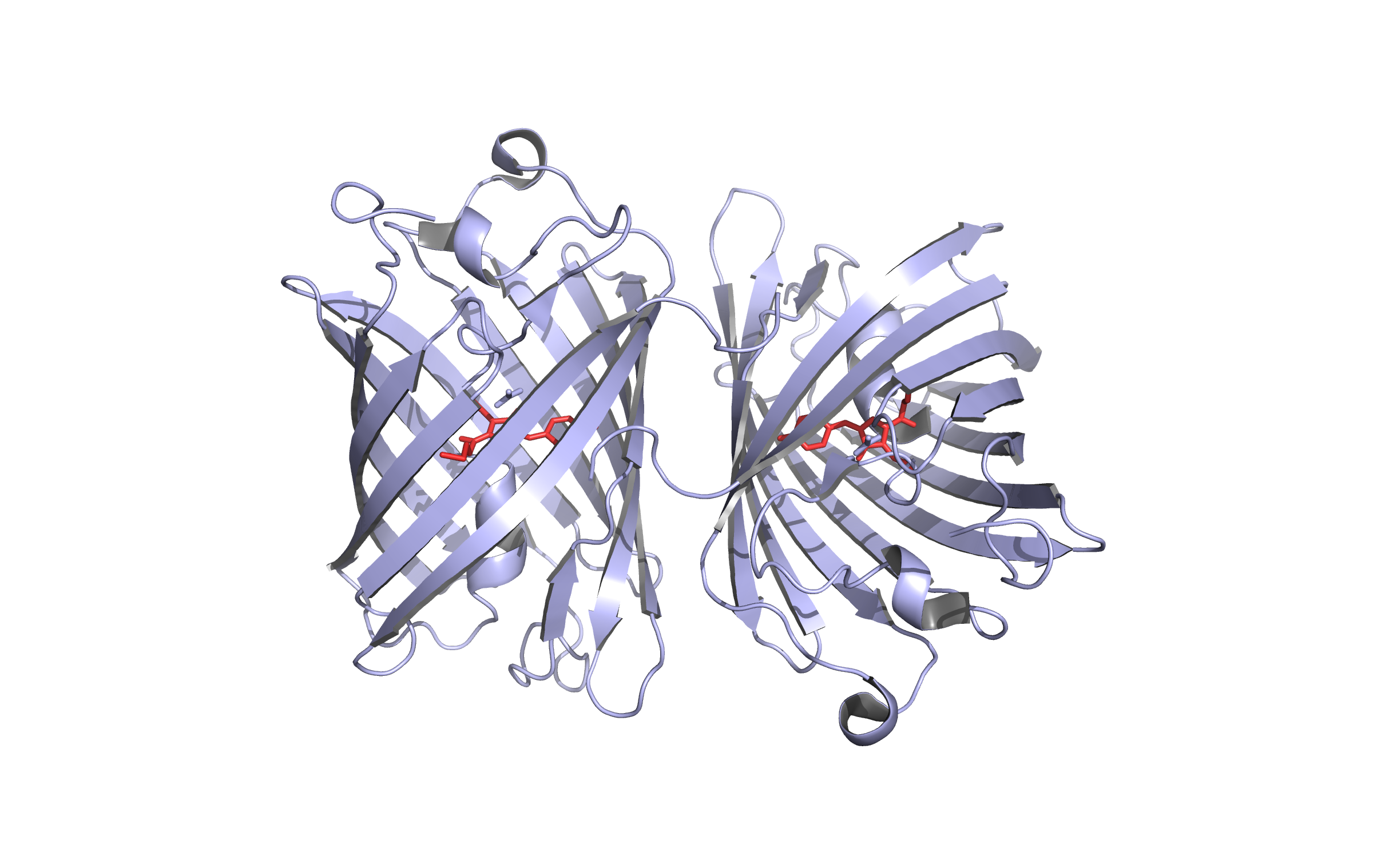
LssmOrange - first modelling
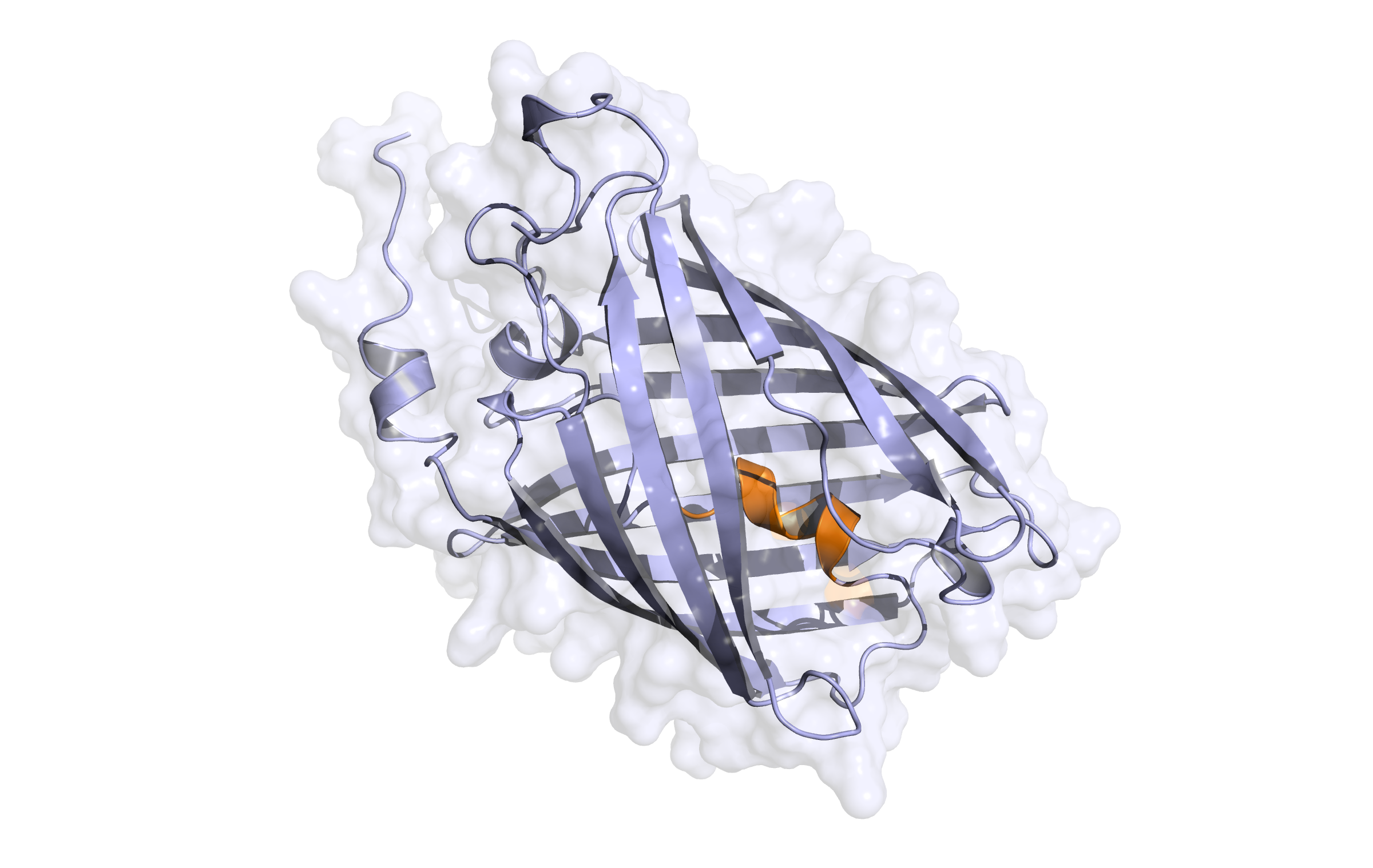

Here we show our hybrid homology model of our protein,
LssmOrange
in ribbon representation. Furthermore we illustrate the average quality z-score as a function of residue number. Nevertheless it was subjected to a final round of simulated annealing minimization in explicit solvent and obtained the following quality Z-scores: