 "
"
Team:KU Leuven/Project/Oscillator/Modelling
From 2013.igem.org
| Line 218: | Line 218: | ||
<br> | <br> | ||
In the case of metabolites, that are produced by enzymes the rate at full production capacity equals <img src="https://static.igem.org/mediawiki/2013/f/fd/Mod_texteq9.PNG" alt="dZ/dt=β_Z∙enzZ-α_Z∙Z"> which becomes <img src="https://static.igem.org/mediawiki/2013/1/1c/Mod_texteq10.PNG" alt="Z=β_Z∙enzZ/α_Z"> at steady-state. When the enzyme is at its own steady-state concentration during maximal production rate this becomes <img src="https://static.igem.org/mediawiki/2013/f/fd/Mod_texteq11.PNG" alt="Z=β_Z∙(β_enzZ/α_enzZ )/α_Z">. | In the case of metabolites, that are produced by enzymes the rate at full production capacity equals <img src="https://static.igem.org/mediawiki/2013/f/fd/Mod_texteq9.PNG" alt="dZ/dt=β_Z∙enzZ-α_Z∙Z"> which becomes <img src="https://static.igem.org/mediawiki/2013/1/1c/Mod_texteq10.PNG" alt="Z=β_Z∙enzZ/α_Z"> at steady-state. When the enzyme is at its own steady-state concentration during maximal production rate this becomes <img src="https://static.igem.org/mediawiki/2013/f/fd/Mod_texteq11.PNG" alt="Z=β_Z∙(β_enzZ/α_enzZ )/α_Z">. | ||
| - | |||
</p> | </p> | ||
</div> | </div> | ||
</div> | </div> | ||
| + | |||
| + | <div class="row-fluid"> | ||
| + | <div class="span12 white"> | ||
| + | <p align = "justify"> | ||
| + | The maximal production rates (β) of the proteins can vary enormously, for instance due to differences in copy number. However, the goal of this oscillator is to have the components in the chromosomal DNA of the cells. We thus assume values that are in the rage of production rates of chromosomal genes. For those ranges we use the assumption that the steady-state concentrations of proteins is equal to β/α (see <a href="#Box2 | Steady-state concentrations>Box 2</a>), we used the α’s as determined above and we use protein abundances as determined by Ishihama <i>et al.</i> (2008). They determined a steady-state concentration for enzymes to be around 65 per cell and for components of the biosynthetic machinery to be around 105. An important remark is that the assumption that steady-state concentrations are equal to β/α is only correct for as long as the production is fully on and not when there were some inhibitory effects active. This means that the lowest protein abundances that are determined by Ishihama <i>et al.</i> can be a lot lower than β/α due to for instance regulation of the production rate, which was consequently not at its maximum. When the production rate is only half of its maximum for instance, the steady-state concentration of the protein is only equal to 0.5∙β/α instead of β/α, which means the lowest values determined by Ishihama <i>et al.</i> be too low. On the other hand the highest values might also be too high, since we are not dealing with components of the biosynthetic machinery. It thus seems wiser to us to discard the extremes and only probe between 6.5∙10^2 and 10^4. We will use these values and transform them to production rates by using the formula from Box 1 and the degradation rates from Table 1. The calculated values can be found in Table 1. | ||
| + | <br> | ||
| + | The maximum production rates due to the enzymes cannot be determined this way, but also span an enormous range. Shaefer <i>et al.</i> (1996) found proof, however, that enzymes that produce quorum-sensing molecules have a low maximum production rate, and they found specifically for the Vibrio fischeri LuxI protein that β equals 1.1 molecule per minute. Since our model is not confined to one enzyme, we will again test a bigger scope. We will again use a range of two decades for this parameter, as can be seen in Table 1. | ||
| + | <br> | ||
| + | The final parameters we will look into is the threshold value. Our method will consist of randomly picking production rates degradation rates of metabolites. As mentioned in <a href="#Box2 | Steady-state concentrations>Box 2</a> the steady-state concentration at full production capacity equals β/α for proteins and <src ="https://static.igem.org/mediawiki/2013/3/3d/Mod_texteq12.PNG" alt="β_Z∙(β_enzZ/α_enzZ )/α_Z">, which means there is no possibility of having a higher level of that protein or metabolite. For the proteins we will use this as an upper bound for setting the possible threshold, since the threshold otherwise would never be reached. We will assume the threshold is reached somewhere in the process, but this is not necessary to have production as long as the Hill coefficient is smaller than infinity. As a lower bound we will allow one decade of variance. The formulas used are displayed in Table 1. The steady-state concentration at full production capacity of metabolites can however vary significantly from the thresholds of the proteins that act as transcription factors. Since the threshold is in reality a function of the interaction between promoter and transcription factor and not of the steady-state concentration of the transcription factor, it would be unfair to use such different values for thresholds for proteins on the one hand and thresholds for metabolites on the other. Instead we will use an the median of all β/α of the proteins for the thresholds for metabolites. Since we use a uniform probability distribution for both α and β the median will be the <img src="https://2013.igem.org/File:Mod_texteq13.PNG" alt="(β_ub+β_lb)/(α_ub+α_lb )">. The resulting value is shown in Table 1, with a lower bound that is one decade lower. | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| + | |||
| + | |||
</body> | </body> | ||
Revision as of 13:22, 27 October 2013



Secret garden
Congratulations! You've found our secret garden! Follow the instructions below and win a great prize at the World jamboree!
- A video shows that two of our team members are having great fun at our favourite company. Do you know the name of the second member that appears in the video?
- For one of our models we had to do very extensive computations. To prevent our own computers from overheating and to keep the temperature in our iGEM room at a normal level, we used a supercomputer. Which centre maintains this supercomputer? (Dutch abbreviation)
- We organised a symposium with a debate, some seminars and 2 iGEM project presentations. An iGEM team came all the way from the Netherlands to present their project. What is the name of their city?
Now put all of these in this URL:https://2013.igem.org/Team:KU_Leuven/(firstname)(abbreviation)(city), (loose the brackets and put everything in lowercase) and follow the very last instruction to get your special jamboree prize!


Oscillator: Model
In this part of the wiki we will describe how we performed an analysis of our proposed oscillating system and the results. This text starts with a small introduction on what we want to achieve with this oscillator, a topic that is more thoroughly elaborated on the design page. Before we started the analysis that is stated here, we looked up how similar networks have been analyzed before in order to see what direction we will take. A full-scale analysis would go beyond the scope of the project so we will stick to an elaborate indicative study. The first step is to translate our network into ODE’s (ordinary differential equations), which we will make more realistic step by step. We will use these to see how easily sustained oscillations form. This is of course not the most impressive feature since there are many known networks that easily produce oscillations. We chose not to include the effect on amplitude and frequency, since that would make the scope of this study explode and the many assumptions we have to make, render it unrealistic. The important feature of our network is its synchronization features. In order to check whether our model achieves rapid resynchronization, we will solve systems of PDE’s (partial differential equations). Since those are a lot more computationally intense, we used the Flemish Super Computer Centre (VSC) in order to do our computations time-efficiently. The explanation of how the network functions and how it attains synchronized oscillations can be found on the page explaining the design. For the transformation into a biological network, we refer you to the wetlab page.
1.1 Exploring our possibilities
Firstly, it is important to clarify what we exactly mean with ‘parameter’. A biochemical system has a very high variability in ranges for transcription rates, translation rates, degradation rates, etc. On top of that these parameters are not perfectly quantified and are subject to changes in the conditions. We checked what part of the parameter space (Box 1) creates oscillations. There are possibilities for doing this in a purely mathematical manner. Tyson (2002) gives a good example of how to study systems that produce biochemical oscillations. Polynikis, Hogan and Bernardo (2009) described modelling approaches for gene regulatory networks more generally. This is typically done by investigating the eigenvalues of the Jacobian matrix of the system. This becomes increasingly more difficult when the number of parameters and variables increases. A high level of non-linearity complicates the study of the behaviour even further. In order to see what is possible we contacted Professor Dirk Roose, an expert in non-linear systems analysis. We explained him we want to investigate what parameter values create a synchronized oscillation. However, our parameter space consists of about 20 parameters that can each vary with more than a factor ten. On top of that we will have highly non-linear equations. Professor Roose told us this amount of variability would make a clean-cut mathematical examination of our model impossible . Since it is not possible to reduce our parameter space, without diverting from our goal of fully studying our system, we decided to use another strategy. We will study this enormous parameter space by generating random sets of parameters throughout this space. This offers a less theoretical, nonetheless effective means of assuring this model robustly produces oscillations.
Box1 | Parameter Space
We will explain this concept with a simple example. Let’s say we want an iGEM team (our system) to develop an oscillator. Two parameters of our system will determine whether this endeavour is successful. These parameters are the number of engineers in a team and the amount of coffee they consume per engineer. As to determine the parameter space we have to find relevant ranges for each of the parameters. We assume more than 10 engineers in the iGEM team is unlikely and a coffee consumption of more than 1.2 liter per day per engineer can also be discarded. The resulting parameter space for this two-parameter model can be easily visualised by a rectangle. This resulting parameter space can be probed and the parameter values we obtained were then used in a simulation which gave as an output whether an oscillator is produced or not. In the figure below we show the result of such a random probing of the parameter space. The blue dots represent unsuccessful endeavours and the orange dots successful ones.
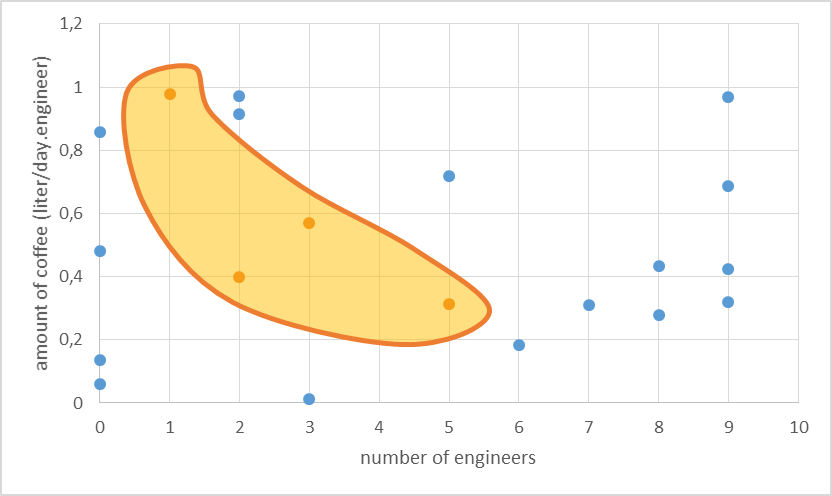 The results allow us to hypothesise on what is necessary to produce an oscillator. Apparently a higher amount of engineers still makes success possible, but only when the coffee consumption is not too high. We hypothesise here that this might be because the engineers get too much energy and start to fight amongst themselves.
We used a similar approach in the testing of our model, with some distinctions though. We deme parameter sets positive when synchronized oscillations take place. But we have 26 parameters and that makes it impossible to visualise our positive parameter space. So we chose another way to look at the parameter space and look at the total fraction of the parameter space that exhibits the wanted behaviour.
The results allow us to hypothesise on what is necessary to produce an oscillator. Apparently a higher amount of engineers still makes success possible, but only when the coffee consumption is not too high. We hypothesise here that this might be because the engineers get too much energy and start to fight amongst themselves.
We used a similar approach in the testing of our model, with some distinctions though. We deme parameter sets positive when synchronized oscillations take place. But we have 26 parameters and that makes it impossible to visualise our positive parameter space. So we chose another way to look at the parameter space and look at the total fraction of the parameter space that exhibits the wanted behaviour.
1.2 Derivation of the ordinary differential equations (ODE’s)
1.2.1 The preliminary system
The logical circuit of our oscillator is displayed in Figure 1 and for an elaborate explanation on this design we refer you to the design page. We will start with a preliminary translation of this network into a system of ODE’s, which is the most used method for modeling gene regulatory networks (Polynikis, Hogan and di Bernardo, 2009). An ODE cannot account for the fact that in reality each of the steps (like transcription and translation) take a finite amount of time. Danino et al. (2010) used delayed differential equations (DDE’s) as a solution for this problem but this complicates the mathematics enormously, especially when we want to take all our parameters into account. We will stick to ODE’s, since that should suffice for the study of our model. The extra delay should benefit the occurrence of oscillations because it helps diminishing peak overlap. The system of ODE’s is created by composing a rate equation for each of the six components of this preliminary circuit Figure 1. Those rate equations give the change of the components over time as a function of the amount of each of the components. The resulting system is displayed as Equation 1. Below we explain the meaning of the different components and afterwards we will explain each of the equations in this system. Further on we will derive the ODE systems for circuits that are slightly altered; by adding the fact that the production of quorum-sensing molecules goes through an enzymatic step and by using the implemented version of the OR gate.
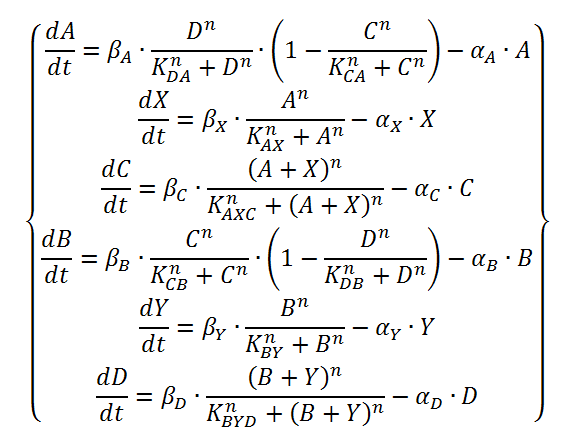 Equation 1: preliminary ODE system exhibiting the behaviour of the oscillator.
Equation 1: preliminary ODE system exhibiting the behaviour of the oscillator.
The functions of the form 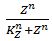 are called Hill functions and are often used to represent unknown regulatory functions (Rosenfeld et al., 2005). With a high Hill coefficient, represented by n in the equations above, this function approximates a step function, as can be seen in Figure 2. The other parameter of the Hill function, indicated by K, gives the moment at which half of the maximum is reached. In the ideal case of a step function, this K value gives the threshold, above which the Hill function returns 1 and below that value, the Hill function returns 0.
are called Hill functions and are often used to represent unknown regulatory functions (Rosenfeld et al., 2005). With a high Hill coefficient, represented by n in the equations above, this function approximates a step function, as can be seen in Figure 2. The other parameter of the Hill function, indicated by K, gives the moment at which half of the maximum is reached. In the ideal case of a step function, this K value gives the threshold, above which the Hill function returns 1 and below that value, the Hill function returns 0.
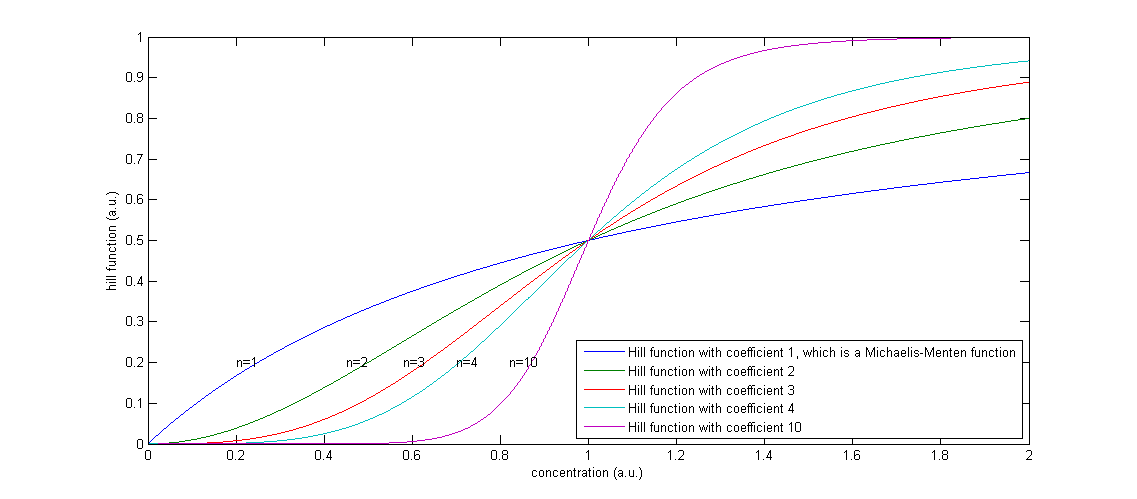 Figure 2: The effect of a higher Hill coefficient
Figure 2: The effect of a higher Hill coefficient
The other parameters in this model are the maximal production rates, which are indicated by β’s and the decay rates, which are indicated by α’s. The degradation term is proportional to the concentration of that component, in which the proportionality constant α can be seen as the percentage of the present proteins that degrades in one unit of time.
We will now discuss the different rate equations and how they are linked to the logical circuit of Figure 1. The concentration of each of the components changes because of a production and a degradation term. The production depends on the presence of the other components and the degradation is as described above. Specifically for A the production is activated in the presence of D and repressed in the presence of C. The logical AND gate can be seen as if both components have to give a positive signal (presence of D and absence of C) in order to have production of A. The format 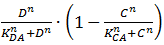 exhibits this behaviour; when C is above its threshold the second term equals zero, when D is absent the first term gives zero. Consequently both D has to be present and C has to be absent to have a non-zero production term. X is a simpler case, since its production is only influenced by A; when there is a sufficient amount of A, there is production of X. C is subject to an OR gate and in order to model this we use
exhibits this behaviour; when C is above its threshold the second term equals zero, when D is absent the first term gives zero. Consequently both D has to be present and C has to be absent to have a non-zero production term. X is a simpler case, since its production is only influenced by A; when there is a sufficient amount of A, there is production of X. C is subject to an OR gate and in order to model this we use 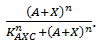 . When either A or X (or a combination of the two) reaches the required level there is production of C. For the other three rate equations the same explanation holds since it is a symmetric system.
. When either A or X (or a combination of the two) reaches the required level there is production of C. For the other three rate equations the same explanation holds since it is a symmetric system.
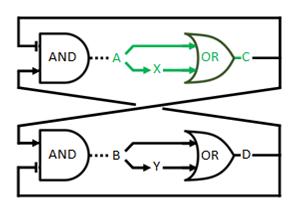
Figure 1: Logical circuit displaying our oscillator
1.2.2 Enzymatic production of quorum-sensing molecules
As mentioned on the design page the quorum-sensing molecules are produced by an enzymatic step. Equation 2 shows the equations that result in that behaviour. The transcription factors control the production of the enzymes rather than A and B directly. The production of A and B is modelled by using Michaelis-Menten kinetics (SA/(KA+SA)). To not overly complicate our model we will assume that the substrates SA and SB are at saturation levels. This means they are a lot higher than KA, KB respectively and the Michaelis-Menten consequently returns 1. This resulting system of equations is shown in Equation 3.
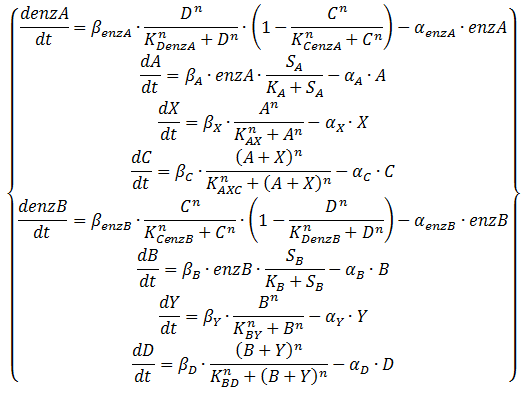
Equation 2: ODE system exhibiting the behaviour of the oscillator, including an enzymatic production step for the QS molecules
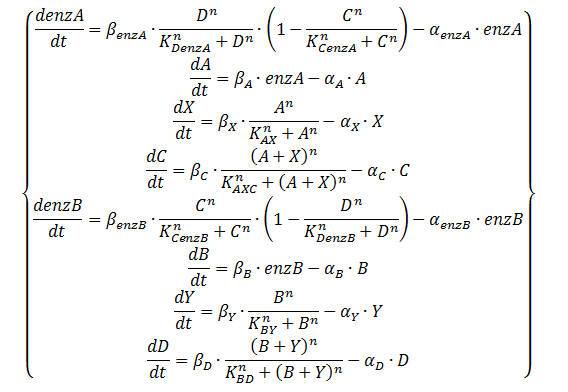
Equation 3: ODE system exhibiting the behaviour of the oscillator, including an enzymatic production step for the QS molecules with the assumption of saturation of those enzymes
1.2.3 Alternative OR gate
In the part before we used an actual OR gate, however, in the wetlab we use an approximation of a real OR gate. We implement the gene for production of C (D) twice, once with a promoter that is induced by A (B) and once with one that is induced by X (Y). This is easier to implement although the behaviour is somewhat different. There is now a higher expression when both the inducers are present. There is no difference in the fact that this displays a asymmetric time delay, of which the importance is discussed in the design page. This altered circuit is displayed as Figure 3.
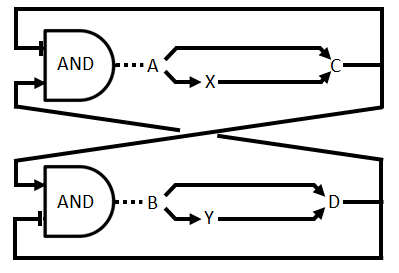
Figure 3: Logical circuit displaying the our oscillator with enzymatic production of the QS molecules and the alternative OR gate
This is translated into Equation 4 by using the format 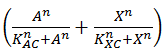 instead of . It is clear that this will return 1 in the case that either A or X are present, 0 when both are absent and 2 when both are present.
instead of . It is clear that this will return 1 in the case that either A or X are present, 0 when both are absent and 2 when both are present.
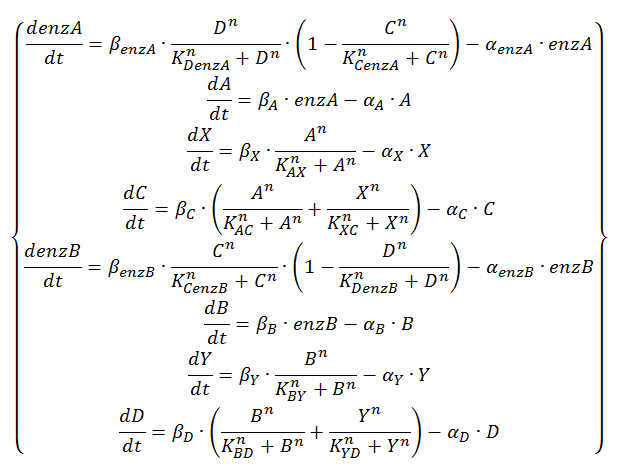
Equation 4: ODE system exhibiting the behavior of the oscillator with enzymatic production of QS molecules at substrate saturation and the alternative OR gate
1.3 Matlab analysis
First we determined the ranges to which the parameters belong. This was done for the degradation rates (α), the production rates (β) and the thresholds (K). We chose random sets of parameters in these ranges and tested whether sustained oscillations take place. Because of the enormous parameter space we could not scan it entirely, but this method still proves whether the tested system inherently produces sustained oscillations. To conduct our computations we made use of the Flemish Super Computer Centre (VSC), which is a partnership of the five Flemish university associations. To use this we first had to rewrite the tests so they could be run in parallel on the supercomputer cluster at our university. Although this seemed straightforward, we encountered an obstacle in the generation of random numbers. When testing on the supercomputer, it appeared like we got 100 identical results in one test. The fault was rapidly determined: the way random number are generated in Matlab. By default, the seed Matlab uses to generate (pseudo)random numbers is always the same if this is the first command you execute. To solve this, we implemented a solution found on (http://www.walkingrandomly.com/?p=2755). This involved getting random numbers out of the Linux kernel random number generator and using these as a seed for the random number generator in Matlab. We tested several systems this way, starting with the simplest and building up to the final one. For each of these systems we checked how many of the tested sets of parameters produce oscillations. We used this to calculate the fraction of oscillating sets in the tested parameter space (see Box 1).
1.3.1 Selection of the parameter range
For the degradation rates of the proteins we assume they follow the N-end rule and are not in one of the unstable categories. This means the dilution rate controls the effective degradation rate and thus we use values that are appropriate for that. More information on how these values have been found can be found on the methyl salicylate modelling page. The ranges can be found in Table 1.
For the colony-wide molecules this approach for the degradation rate cannot be used, since they are present inside as well as outside of the cells. Those molecules thus become diluted when the volume of the colony increases, but not per se when the amount of cells increases as is the case for stable proteins. When we think about using quorum sensing molecules, the colony-wide molecules are metabolites. A potential problem might then be the slow degradation rate as encountered by Danino et al. (2010). On the wetlab page, we describe the use of an actively degrading enzyme as a solution for this issue of slow degradation. Since our model does not require a specific chemical species for oscillating, we will allow a broader spectrum of possibilities than through exactly that degrading enzyme. We will simplify the model by assuming a uniform degradation throughout the colony, but by allowing two decades of variance, we will still allow for a broad spectrum of possibilities. We will use the degradation (dilution) rate of the proteins as a mark for the degradation rate of the colony-wide molecules, since the volume of a colony also increases when the amount of cells augments. The two decades of variance in the degradation rate of colony-wide molecules will range from one tenth of the degradation rate used for proteins to ten times that degradation rate. Allowing values lower than the degradation rate used for proteins is based on the fact that the growth of the volume of a colony is slower than the growth of the number of cells. Allowing values higher than the degradation rate used for proteins is based on the possibility that the colony-wide molecules are for instance actively degraded by some produced enzymes or a fast spontaneous degradation. These values can also be seen in Table 1.
Box2 | Steady-state concentrations
A steady-state concentration of proteins can be easily determined by setting each of the rates to zero. In the case of maximal production rate, the rate equation has the following form: 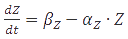 and when
and when 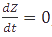 , the equation can be easily converted to
, the equation can be easily converted to 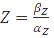 , which gives the steady-state concentration of Z, or
, which gives the steady-state concentration of Z, or 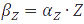 , when all but the production rates are known.
, when all but the production rates are known.
In the case of metabolites, that are produced by enzymes the rate at full production capacity equals 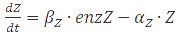 which becomes
which becomes 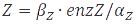 at steady-state. When the enzyme is at its own steady-state concentration during maximal production rate this becomes
at steady-state. When the enzyme is at its own steady-state concentration during maximal production rate this becomes 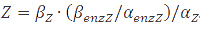 .
.
The maximal production rates (β) of the proteins can vary enormously, for instance due to differences in copy number. However, the goal of this oscillator is to have the components in the chromosomal DNA of the cells. We thus assume values that are in the rage of production rates of chromosomal genes. For those ranges we use the assumption that the steady-state concentrations of proteins is equal to β/α (see Box 2 the steady-state concentration at full production capacity equals β/α for proteins and  . The resulting value is shown in Table 1, with a lower bound that is one decade lower.
. The resulting value is shown in Table 1, with a lower bound that is one decade lower.