 "
"
Team:TU Darmstadt/modelling/Structure
From 2013.igem.org
| Line 111: | Line 111: | ||
} | } | ||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| Line 195: | Line 186: | ||
padding: 5px 5px 5px 5px; | padding: 5px 5px 5px 5px; | ||
font-size: 100%; | font-size: 100%; | ||
| + | } | ||
| + | LI.list1 {list-style: none; color:white;} | ||
| + | .blacktext {color:black} | ||
| + | p, ul { | ||
| + | padding: 0; | ||
| + | margin: 0; | ||
} | } | ||
| Line 277: | Line 274: | ||
| - | + | ||
<br> | <br> | ||
| Line 283: | Line 280: | ||
<!-- Überschrifft --> | <!-- Überschrifft --> | ||
<center> | <center> | ||
| - | + | ||
<br> | <br> | ||
<br> | <br> | ||
| Line 295: | Line 292: | ||
<font size="3" color="#F0F8FF" face="Arial regular"> | <font size="3" color="#F0F8FF" face="Arial regular"> | ||
<p text-aligne:left style="margin-left:50px; margin-right:50px" align="left"> | <p text-aligne:left style="margin-left:50px; margin-right:50px" align="left"> | ||
| - | Description how our Yasara script calculates homology model[7]: | + | Description how our Yasara script calculates homology model[7]:<br> |
<img alt="DKL" src="https://static.igem.org/mediawiki/2012/5/5e/Aln%2BpnB.png" width="400" height="216,5" align="right"> | <img alt="DKL" src="https://static.igem.org/mediawiki/2012/5/5e/Aln%2BpnB.png" width="400" height="216,5" align="right"> | ||
| - | |||
| Line 315: | Line 311: | ||
</ol> | </ol> | ||
| - | |||
| - | |||
<!-- Überschrifft --> | <!-- Überschrifft --> | ||
<center> | <center> | ||
| - | + | ||
<br> | <br> | ||
<br> | <br> | ||
| Line 355: | Line 349: | ||
| - | + | ||
<br> | <br> | ||
<!-- Überschrifft --> | <!-- Überschrifft --> | ||
<center> | <center> | ||
| - | + | ||
<br> | <br> | ||
<br> | <br> | ||
| Line 369: | Line 363: | ||
</center> | </center> | ||
| - | |||
| - | + | ||
<font size="3" color="#F0F8FF" face="Arial regular"> | <font size="3" color="#F0F8FF" face="Arial regular"> | ||
<p text-aligne:center style="margin-left:50px; margin-right:50px"> | <p text-aligne:center style="margin-left:50px; margin-right:50px"> | ||
| Line 387: | Line 380: | ||
<font size="5" color="#F0F8FF" face="Arial regular">First model LssmOrange</font> | <font size="5" color="#F0F8FF" face="Arial regular">First model LssmOrange</font> | ||
</p> | </p> | ||
| - | + | ||
| - | + | ||
<font size="3" color="#F0F8FF" face="Arial regular"> | <font size="3" color="#F0F8FF" face="Arial regular"> | ||
<p text-aligne:center style="margin-left:50px; margin-right:50px"> | <p text-aligne:center style="margin-left:50px; margin-right:50px"> | ||
| - | |||
This model is a monomer, and based on the following sequence alignment: | This model is a monomer, and based on the following sequence alignment: | ||
| + | <p text-aligne:left style="margin-left:60px; margin-right:60px"><font size="3" color="#F0F8FF" face="Arial regular"> | ||
| + | <div align="left" style="margin-left:30px; margin-right:50px"> | ||
| + | <ol> | ||
| + | <li class=list1><b>SecStr:</b></li> | ||
| + | <li class=list1 style="max-width:1000px; word-wrap:break-word;"> CCCCCCCCCCCCCCCCEEEEEEEEEEECCEEEEEEEEEEECCCCCEEEEEEEEEECCCCCCCCCHHHCCCCCCCCCCCCCCCCCHHHHHHCCCCCEEEEEEEEEECCCEEEEEEEEEEECCEEEEEEEEEEECCCCCCCCCCCCCCCCCCEEEEEEEECCEEEEEEEEEEEECCCCEEEEEEEEEEECCCCCCCCCCEEEEEEEEEEEECCCCCEEEEEEEEEEECCCCCCCCCCC</li> | ||
| + | <li class=list1><b>Target:</b></li> | ||
| + | <li class=list1 style="max-width:1000px; word-wrap:break-word;"> MVSKGEENNMAIIKEFMRFKVRMEGSVNGHEFEIEGEGEGRPYEGFQTVKLKVTKGGPLPFAWDILSPQFTYGSKAYVKHPADIPDYLKLSFPEGFKWERVMNFEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMGMEASSERMYPEDGALKGEDKLRLKLKDGGHYTSEVKTTYKAKKPVQLPGAYIVDIKLDITSHNEDYTIVEQYERAEGRHSTGGMDELYK</li> | ||
| + | <li class=list1><b>Match:</b></li> | ||
| + | <li class=list1 style="max-width:1000px; word-wrap:break-word;"> EE:NMAIIKEFMRFK::MEGSVNGHEFEIEGEGEGRPYEG QT:KLKVTKGGPLPFAWDILSPQF SKAYVKHPADIPDYLKLSFPEGFKWERVMNFEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMG EA SERMYPEDGALKGE K|RLKLKDGGHY :EVKTTYKAKKPVQLPGAY :: KLDITSHNEDYTIVEQYER EGRHSTGGMDELYK</li> | ||
| + | <li class=list1><b>Template:</b></li> | ||
| + | <li class=list1 style="max-width:1000px; word-wrap:break-word;"> .....EEDNMAIIKEFMRFKTHMEGSVNGHEFEIEGEGEGRPYEGTQTAKLKVTKGGPLPFAWDILSPQF...SKAYVKHPADIPDYLKLSFPEGFKWERVMNFEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMGWEACSERMYPEDGALKGEMKMRLKLKDGGHYDAEVKTTYKAKKPVQLPGAYNTNTKLDITSHNEDYTIVEQYERNEGRHSTGGMDELYK</li> | ||
| + | <li class=list1><b>SecStr:</b></li> | ||
| + | <li class=list1 style="max-width:1000px; word-wrap:break-word;"> ....HHHHHHCCCCCEEEEEEEEEEETTEEEEEEEEEEEETTTEEEEEEEEEEECCCCCCGGGGGGGGG...TTTTCECTTTTCHHHHHHCCCEEEEEEEEEETTEEEEEEEEEEEEETTEEEEEEEEEEECCTTTTCTTTTCCEEEECEEEEEEEETTEEEEEEEEEEEETTEEEEEEEEEEEEEECCCCCCCCCEEEEEEEEEEEEETTTEEEEEEEEEEEECCCCTTTTCCC/li> | ||
| + | </ol> | ||
| + | </div> | ||
| + | </font></p> | ||
| + | |||
| + | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
| - | |||
<font size="3" color="#F0F8FF" face="Arial regular"> | <font size="3" color="#F0F8FF" face="Arial regular"> | ||
<p text-aligne:center style="margin-left:50px; margin-right:50px"> | <p text-aligne:center style="margin-left:50px; margin-right:50px"> | ||
| Line 431: | Line 429: | ||
<!-- Tabelle --> | <!-- Tabelle --> | ||
| - | + | ||
<br> | <br> | ||
<br> | <br> | ||
| - | + | ||
| - | + | ||
| - | <table border="1"> | + | <table border="1" style="background-color: transparent;"> |
<tr> | <tr> | ||
<th><b>Check Type</b></th> | <th><b>Check Type</b></th> | ||
| Line 472: | Line 470: | ||
</table> | </table> | ||
| - | + | ||
<br> | <br> | ||
| Line 480: | Line 478: | ||
<br> | <br> | ||
| - | |||
<p text-aligne:left style="margin-left:50px; margin-right:50px"> | <p text-aligne:left style="margin-left:50px; margin-right:50px"> | ||
<font size="5" color="#F0F8FF" face="Arial regular">LssmOrange - second modelling</font> | <font size="5" color="#F0F8FF" face="Arial regular">LssmOrange - second modelling</font> | ||
</p> | </p> | ||
| - | |||
| - | + | ||
<font size="3" color="#F0F8FF" face="Arial regular"> | <font size="3" color="#F0F8FF" face="Arial regular"> | ||
<p text-aligne:center style="margin-left:50px; margin-right:50px"> | <p text-aligne:center style="margin-left:50px; margin-right:50px"> | ||
| Line 496: | Line 492: | ||
<pre>SecStr: CCCCCCCCCCCCCCCCEEEEEEEEEEECCEEEEEEEEEEECCCCCEEEEEEEEEECCCCCCCCCHHHCCCCCCCCCCCCCCCCCHHHHHHCCCCCEEEEEEEEEECCCEEEEEEEEEEECCEEEEEEEEEEECCCCCCCCCCCCCCCCCCEEEEEEEECCEEEEEEEEEEEECCCCEEEEEEEEEEECCCCCCCCCCEEEEEEEEEEEECCCCCEEEEEEEEEEECCCCCCCCCCC<br>Target: MVSKGEENNMAIIKEFMRFKVRMEGSVNGHEFEIEGEGEGRPYEGFQTVKLKVTKGGPLPFAWDILSPQFTYGSKAYVKHPADIPDYLKLSFPEGFKWERVMNFEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMGMEASSERMYPEDGALKGEDKLRLKLKDGGHYTSEVKTTYKAKKPVQLPGAYIVDIKLDITSHNEDYTIVEQYERAEGRHSTGGMDELYK<br>Match: AIIKEFMRFKV:MEGSVNGH FEIEGEGEGRPYEG QT:KLKVTKGGPLPF:WDILSPQF S:AYVKHPADIPDY:KLSFPEGFKWERVM:FEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMG EA SERMYPEDGALKGE K R:KLKDGGHY :EVKTTYKAKKPVQLPGAY V: KLDITSHNEDYTIVEQYERAEGRHS <br>Template:..........AIIKEFMRFKVHMEGSVNGHVFEIEGEGEGRPYEGTQTAKLKVTKGGPLPFTWDILSPQF...SNAYVKHPADIPDYFKLSFPEGFKWERVMKFEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMGWEALSERMYPEDGALKGEVKPRVKLKDGGHYDAEVKTTYKAKKPVQLPGAYNVNRKLDITSHNEDYTIVEQYERAEGRHS.........<br>SecStr: ..........CCCCCCEEEEEEEEEEETTEEEEEEEEEEEETTTEEEEEEEEEEECCCCCCCCCCGGGGC...TTTTCECTTTTCHHHHHHCCCEEEEEEEEEETTEEEEEEEEEEEEETTEEEEEEEEEEECCTTTTCTTTTCCEEEECEEEEEEEETTEEEEEEEEEEEETTEEEEEEEEEEEEEECCCCCCCCCEEEEEEEEEEEEETTTEEEEEEEEEEEECC.........<br></pre> | <pre>SecStr: CCCCCCCCCCCCCCCCEEEEEEEEEEECCEEEEEEEEEEECCCCCEEEEEEEEEECCCCCCCCCHHHCCCCCCCCCCCCCCCCCHHHHHHCCCCCEEEEEEEEEECCCEEEEEEEEEEECCEEEEEEEEEEECCCCCCCCCCCCCCCCCCEEEEEEEECCEEEEEEEEEEEECCCCEEEEEEEEEEECCCCCCCCCCEEEEEEEEEEEECCCCCEEEEEEEEEEECCCCCCCCCCC<br>Target: MVSKGEENNMAIIKEFMRFKVRMEGSVNGHEFEIEGEGEGRPYEGFQTVKLKVTKGGPLPFAWDILSPQFTYGSKAYVKHPADIPDYLKLSFPEGFKWERVMNFEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMGMEASSERMYPEDGALKGEDKLRLKLKDGGHYTSEVKTTYKAKKPVQLPGAYIVDIKLDITSHNEDYTIVEQYERAEGRHSTGGMDELYK<br>Match: AIIKEFMRFKV:MEGSVNGH FEIEGEGEGRPYEG QT:KLKVTKGGPLPF:WDILSPQF S:AYVKHPADIPDY:KLSFPEGFKWERVM:FEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMG EA SERMYPEDGALKGE K R:KLKDGGHY :EVKTTYKAKKPVQLPGAY V: KLDITSHNEDYTIVEQYERAEGRHS <br>Template:..........AIIKEFMRFKVHMEGSVNGHVFEIEGEGEGRPYEGTQTAKLKVTKGGPLPFTWDILSPQF...SNAYVKHPADIPDYFKLSFPEGFKWERVMKFEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMGWEALSERMYPEDGALKGEVKPRVKLKDGGHYDAEVKTTYKAKKPVQLPGAYNVNRKLDITSHNEDYTIVEQYERAEGRHS.........<br>SecStr: ..........CCCCCCEEEEEEEEEEETTEEEEEEEEEEEETTTEEEEEEEEEEECCCCCCCCCCGGGGC...TTTTCECTTTTCHHHHHHCCCEEEEEEEEEETTEEEEEEEEEEEEETTEEEEEEEEEEECCTTTTCTTTTCCEEEECEEEEEEEETTEEEEEEEEEEEETTEEEEEEEEEEEEEECCCCCCCCCEEEEEEEEEEEEETTTEEEEEEEEEEEECC.........<br></pre> | ||
--> | --> | ||
| - | + | ||
<font size="3" color="#F0F8FF" face="Arial regular"> | <font size="3" color="#F0F8FF" face="Arial regular"> | ||
<p text-aligne:center style="margin-left:50px; margin-right:50px"> | <p text-aligne:center style="margin-left:50px; margin-right:50px"> | ||
In the alignment, 214 of 236 target residues (90.7%) are aligned to template residues. Among these aligned residues, the sequence identity is 91.6% and the sequence similarity is 93.0% ('similar' means that the BLOSUM62 score is > 0). | In the alignment, 214 of 236 target residues (90.7%) are aligned to template residues. Among these aligned residues, the sequence identity is 91.6% and the sequence similarity is 93.0% ('similar' means that the BLOSUM62 score is > 0). | ||
| - | + | ||
<br> | <br> | ||
<br> | <br> | ||
| Line 524: | Line 520: | ||
<br> | <br> | ||
| - | + | ||
| - | + | ||
<table border="1"> | <table border="1"> | ||
<tr> | <tr> | ||
| Line 560: | Line 555: | ||
</table> | </table> | ||
| - | + | ||
| Line 568: | Line 563: | ||
<br> | <br> | ||
| - | + | ||
<p text-aligne:left style="margin-left:50px; margin-right:50px"> | <p text-aligne:left style="margin-left:50px; margin-right:50px"> | ||
<font size="5" color="#F0F8FF" face="Arial regular">Third model LssmOrange (dimer)</font> | <font size="5" color="#F0F8FF" face="Arial regular">Third model LssmOrange (dimer)</font> | ||
</p> | </p> | ||
| - | + | ||
| - | + | ||
<font size="3" color="#F0F8FF" face="Arial regular"> | <font size="3" color="#F0F8FF" face="Arial regular"> | ||
<p text-aligne:center style="margin-left:50px; margin-right:50px"> | <p text-aligne:center style="margin-left:50px; margin-right:50px"> | ||
| Line 588: | Line 582: | ||
<br> | <br> | ||
| - | <img alt="lssmOrange" src="/wiki/images/e/ed/LssmOrange_third_activeHelix_surface_light2.png" width="75%" height="75% | + | <img align="center" alt="lssmOrange" src="/wiki/images/e/ed/LssmOrange_third_activeHelix_surface_light2.png" width="75%" height="75%" > |
<br> | <br> | ||
| Line 594: | Line 588: | ||
<font size="3" color="#F0F8FF" face="Arial regular"> | <font size="3" color="#F0F8FF" face="Arial regular"> | ||
| - | <p text-aligne:center style="margin-left:50px; margin-right:50px"> | + | <p text-aligne:center style="margin-left:50px; margin-right:50px"> |
<img alt="mod_qual" src="https://static.igem.org/mediawiki/2013/e/ef/Lsso_4kge-~02_quality.png" width="800" height="116" > | <img alt="mod_qual" src="https://static.igem.org/mediawiki/2013/e/ef/Lsso_4kge-~02_quality.png" width="800" height="116" > | ||
| Line 608: | Line 602: | ||
<br> | <br> | ||
| - | + | ||
| - | + | ||
<table border="1"> | <table border="1"> | ||
<tr> | <tr> | ||
| Line 644: | Line 637: | ||
</table> | </table> | ||
| - | + | ||
| Line 689: | Line 682: | ||
</center> | </center> | ||
</body> | </body> | ||
| + | </html> | ||
Revision as of 23:21, 4 October 2013
Homology Modelling
While our proteins are functionally described in literature and during the IGEM competition, only part of the structures are available in the protein data bank. For further work and visualizations, protein structures are indispensable. We used Yasara Structure [1] to calculate 3-dimensional structures of all of our proteins for the IGEM.
Workflow
Description how our Yasara script calculates homology model[7]:

- Sequence is PSI-BLASTed against Uniprot [2]
- Calculation of a position-specific scoring matrix (PSSM) from related sequences
- Using the PSSM to search the PDB for potential modeling templates
- The Templates are ranked based on the alignment score and the structural quality[3]
- Deriving additional information’s for template and target (prediction of secondary structure, structure-based alignment correction by using SSALN scoring matrices [4]).
- A graph of the side-chain rotamer network is built, dead-end elimination is used to find an initial rotamer solution in the context of a simple repulsive energy function [5]
- The loop-network is optimized using a high amount of different orientations
- Side-chain rotamers are fine-tuned considering electrostatic and knowledge-based packing interactions as well as solvation effects.
- An unrestrained high-resolution refinement with explicit solvent molecules is run, using the latest knowledge-based force fields[6].
Application
All these steps are performed to every template used for the modeling approach. For our project we set the maximum amount of templates to 20. Every derived structure is evaluated using an average per-residue quality Z-scores. At last a hybrid model is built containing the best regions of all predictions. This procedure make prediction’s accurate and thus more realistic. For the evaluation we used the Yasara Z-scores.A Z-score describes how many standard deviations the model quality is away from the average high-resolution X-ray structure. Negative values indicate that the homology model looks worse than a high-resolution X-ray structure. The overall Z-scores for all models have been calculated as the weighted averages of the individual Z-scores using the formula Overall = 0.145*Dihedrals + 0.390*Packing1D + 0.465*Packing3D [7].
Parameters
We used the Yasara script hm_build.mcr for the model creation with the following parameters:
- Modeling speed (slow = best): Slow
- Number of PSI-BLAST iterations in template search (PsiBLASTs): 3
- Maximum allowed PSI-BLAST E-value to consider template (EValue Max): 0.5
- Maximum number of templates to be used (Templates Total): 20
- Maximum number of templates with same sequence (Templates SameSeq): 1
- Maximum oligomerization state (OligoState): 4 (tetrameric)
- Maximum number of alignment variations per template: (Alignments): 5
- Maximum number of conformations tried per loop (LoopSamples): 50
- Maximum number of residues added to the termini (TermExtension): 10
Results
Since the target sequence was the only available information, possible templates were identified by running 6 PSI-BLAST iterations to extract a position specific scoring matrix (PSSM) from Uniprot, and then searching the PDB for a match (i.e. hits with an E-value below the homology modeling cutoff 0.5)
We present the best three models from a total of 14 listed in the model ranking and sorted by their overall quality Z-scores.
First model LssmOrange
This model is a monomer, and based on the following sequence alignment:
In the alignment, 228 of 236 target residues (96.6%) are aligned to template residues. Among these aligned residues, the sequence identity is 93.0% and the sequence similarity is 94.7% ('similar' means that the BLOSUM62 score is > 0).
LssmOrange - second modelling
In the alignment, 214 of 236 target residues (90.7%) are aligned to template residues. Among these aligned residues, the sequence identity is 91.6% and the sequence similarity is 93.0% ('similar' means that the BLOSUM62 score is > 0).
Third model LssmOrange (dimer)
In the alignment, 207 of 236 target residues (87.7%) are aligned to template residues. Among these aligned residues, the sequence identity is 58.0% and the sequence similarity is 73.9% ('similar' means that the BLOSUM62 score is > 0)
[1] E. Krieger, G. Koraimann, and G. Vriend, “Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field.,” Proteins, vol. 47, no. 3, pp. 393–402, 2002.
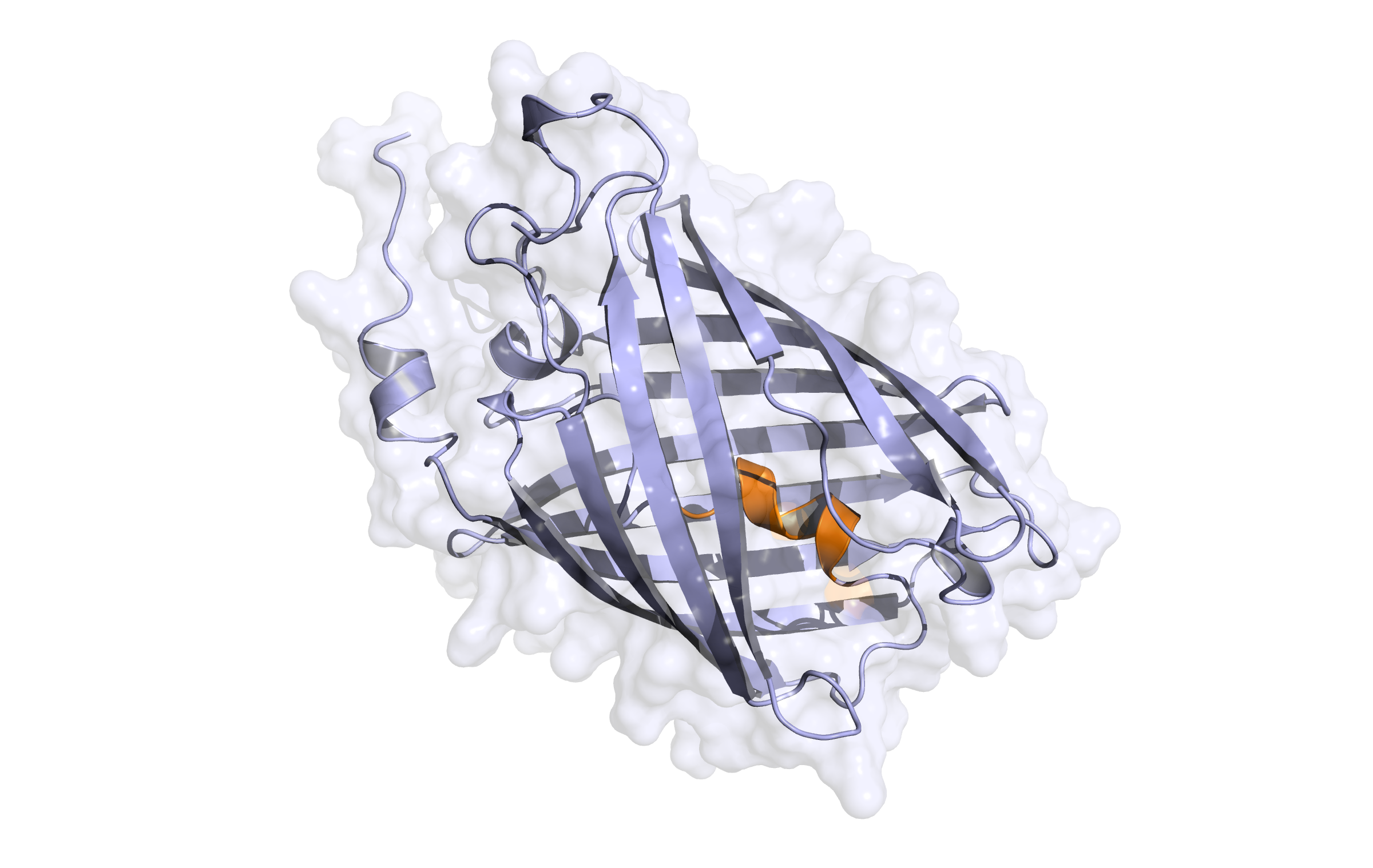

Here, the first hybrid homology model of our protein,
LssmOrange
is shown in ribbon representation. Furthermore, we illustrate the average quality z-score as a function of residue number. Nevertheless, it was subjected to a final round of simulated annealing minimization in explicit solvent and obtained the following quality Z-scores:
Check Type
Quality Z-score
Comment
Dihedrals
2.363
Optimal
Packing 1D
-0.864
Good
Packing 3D
-1.138
Satisfactory
Overall
-0.524
Good
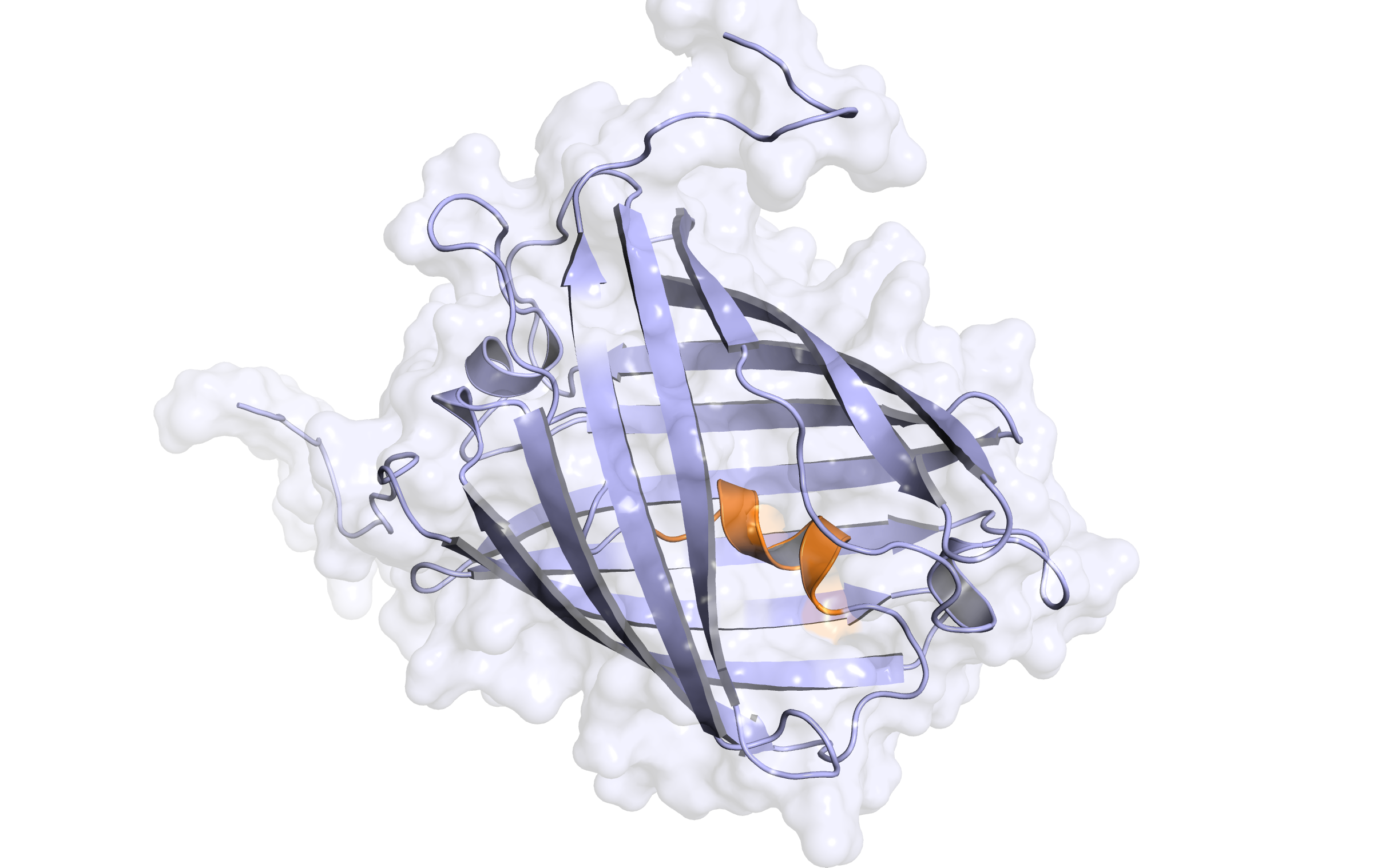

Here, the second hybrid homology model of our protein,
LssmOrange
is shown in ribbon representation. Again, we illustrate the average quality z-score as a function of residue number and obtained the following quality Z-scores:
Check Type
Quality Z-score
Comment
Dihedrals
2.125
Optimal
Packing 1D
-0.936
Good
Packing 3D
-1.259
Satisfactory
Overall
-0.642
Good
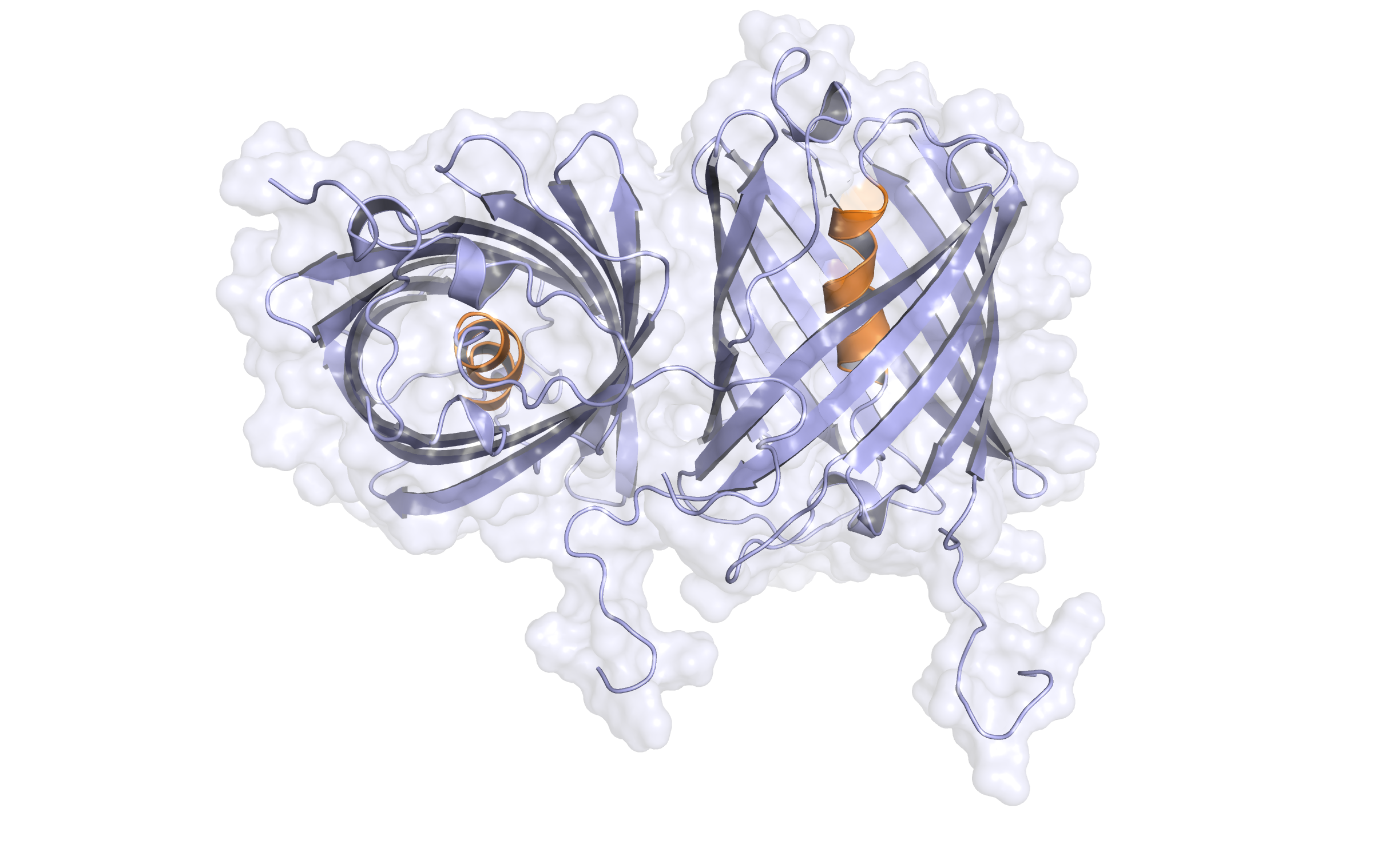

Here, the third hybrid homology model of our protein,
LssmOrange
is shown in ribbon representation. Again, we illustrate the average quality z-score as a function of residue number and obtained the following quality Z-scores:
Check Type
Quality Z-score
Comment
Dihedrals
1.237
Optimal
Packing 1D
-2.083
Poor
Packing 3D
-1.651
Satisfactory
Overall
-1.400
Satisfactory
References
[2] S. F. Altschul, T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman, “Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.,” Nucleic Acids Res, vol. 25, no. 17, pp. 3389–3402, Sep. 1997.
[3] R. W. Hooft, G. Vriend, C. Sander, and E. E. Abola, “Errors in protein structures.,” Nature, vol. 381, no. 6580. Nature Publishing Group, p. 272, 1996.
[4] D. T. Jones, “Protein secondary structure prediction based on position-specific scoring matrices,” Journal of Molecular Biology, vol. 292, no. 2, pp. 195–202, 1999.
[5] A. A. Canutescu, A. A. Shelenkov, and R. L. Dunbrack, “A graph-theory algorithm for rapid protein side-chain prediction.,” Protein Science, vol. 12, no. 9, pp. 2001–2014, 2003.
[6] E. Krieger, K. Joo, J. Lee, J. Lee, S. Raman, J. Thompson, M. Tyka, D. Baker, and K. Karplus, “Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8.,” Proteins, vol. 77 Suppl 9, no. June, pp. 114–122, 2009.
[7] http://www.yasara.org/homologymodeling.htm