 "
"
Team:IIT Madras/DryLabAndModelling
From 2013.igem.org
|
|
|
Dry Lab and Modelling
BACKGROUND
Molecular docking is a widely-used computational tool for the study of molecular recognition, which aims to predict the binding mode and binding affinity of a complex formed by two or more constituent molecules with known structures. Protein-ligand docking in particular, is very important because of its therapeutic applications in modern structure-based drug design.
Our team chose to do docking experiments in order to evaluate the binding characteristics of Shiga toxin and the peptide designed to neutralise it, as it is important to validate the results generated from experimental techniques.
Since only the sequence of the peptide(WHWTWLSEY) was known, it was necessary to model the structure of the peptide. For this purpose, we used the online webserver, PEP-FOLD(Yimin SHEN et al.). PEP-FOLD is based on the concept of structural alphabet (SA) - i.e. a description of apolypeptidic conformation as a series of local canonical conformations, and uses a HMM (Hidden Markov Chain)-derived SA of 27 letters to describe proteins as series of overlapping fragments of four amino acids. PEP-FOLD is based on a two-step procedure:
-
prediction of a limited set of SA letters at each position from peptide amino acid sequence
-
assembly of the prototype fragments associated with each SA letter using
a greedy algorithm and a generic protein coarse-grained force field.
As mentioned earlier, Shiga toxin binds to human Globotriaosylceramide (Gb3Cer) receptors which are present in the endothelium of mammalian cells. The crystal structure of Gb3 bound to Shiga toxin has already been determined by crystallization (PDB id - 1BOS) performed by Ling et.al. This structure was used in our experiments as a reference structure for carrying out docking studies.
Further work done by Miura et.al., on Gb3 mimicking peptides revealed the Molecular dynamics and binding characteristics of the truncated peptide considering only the first five amino residues of our peptide (WHWTWLSEY). In order to compare the docking characteristics of the entire peptide as opposed to the truncated peptide that was used in the publication, we carried out our bioinformatics experiments.
TOOLS: HADDOCK
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an information-driven flexible docking approach for the modeling of biomolecular complexes. HADDOCK can deal with a large class of modelling problems including protein-protein, protein-nucleic acids and protein-ligand complexes. For the purpose of our studies, we used the Easy Interface of the HADDOCK server which requires several parameters including active and passive residues.
Since our peptide (WHWTWLSEY) is a Gb3 receptor mimic, it is reasonable to assume that the residues in Shiga toxin to which the peptide binds are the same ones to which Gb3 binds .
These active residues were obtained from PDB. These were found to be Lys(33), Asn(35),Asp(36), Asp(38), Thr(41), Glu(48), Thr(51), Asn(52), Arg(53), Trp(54), Asn(55), Thr(74), Asn(75), Gly(80), Gly(82), Phe(83).
RESULTS
HADDOCK uses the HADDOCK score to rank the clusters generated during docking. This is a weighted sum of:
-
Electrostatic energy (weight 0.2)
-
Van der Waals energy (weight 1.0)
-
Desolvation energy (weight 1.0)
-
Restraints violation energies (distance, SANI) (weight 0.1)
According to this ranking score, we shortlisted the best models from the top 5 clusters.These were further analysed on the basis of low RMSD to the original structure of Gb3 bound to Shiga toxin.
|
Clusters |
HADDOCK score |
RMSD from structure of lowest energy |
Z Score |
|
1 |
-110.4 +/- 6.1 |
0.4 +/- 0.2 |
-1.5 |
|
2 |
-100.8 +/- 6.2 |
1.6 +/- 0.3 |
-0.9 |
|
3 |
-93.6 +/- 10.2 |
1.0 +/- 0.3 |
-0.5 |
|
4 |
-91.2 +/- 3.9 |
2.4 +/- 0.0 |
-0.3 |
|
5 |
-70.9 +/- 7.4 |
2.2 +/- 0.2 |
0.9 |
|
6 |
-69.0 +/- 4.9 |
2.0 +/- 0.1 |
1 |
|
7 |
-64.2 +/- 14.9 |
2.0 +/- 0.1 |
1.3 |
Cluster 1


Cluster 2

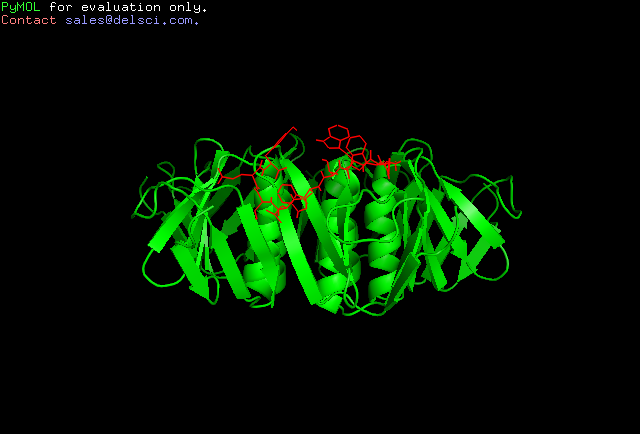
Cluster 3
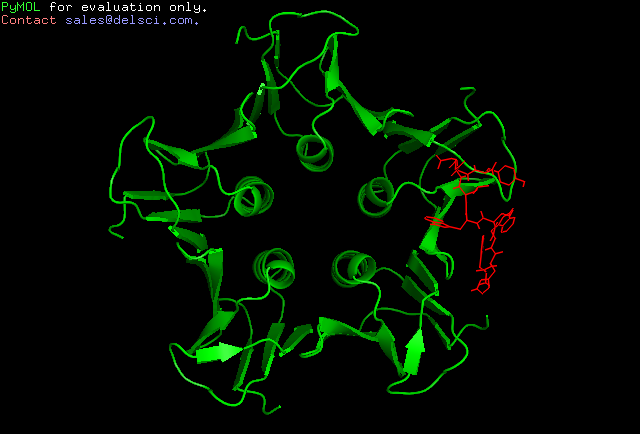

Cluster 4


Cluster 5


Amongst the predicted results, Cluster 2 seems to be most suited as an interacting partner to Shiga toxin. This can be attributed to the optimum energy values assigned by HADDOCK as well as the high similarities of the binding modes of the peptide to the toxin as compared to the same of Gb3-Shiga toxin complex. This was analysed by the RMSD values as indicated by Pymol following structural alignment. The RMSD value obtained was 0.359.
SCOPE FOR FURTHER WORK
Our team intends to construct a peptide library using tools like SDM to create mutants of the peptide and analysing interactions of these with Shiga Toxin. We can also search for sequences homologous to the toxin which could be harmful in nature and test their interactions with peptide library.This is a high-throughput analysis and can be validated further by several wet lab techniques.