|
|
| Line 59: |
Line 59: |
| | '''2. The plasmid that is returned from DNA2.0 will be amplified in E.coli. The following protocol outlines the procedure by which this will take place:''' | | '''2. The plasmid that is returned from DNA2.0 will be amplified in E.coli. The following protocol outlines the procedure by which this will take place:''' |
| | | | |
| - | ===Competent cell preparation=== | + | ===[https://2013.igem.org/Team:Newcastle/Notebook/protocols#Escherichia_coli_Competent_Cell_Preparation Competent cell preparation]=== |
| | | | |
| - | • Inoculate 300ml of LB broth with 1/20 volume of an overnight culture of the desired strain (e.g. 15ml overnight culture for 300ml of LB)
| |
| | | | |
| - | • Grow the cells at 37°C (with shaking) to an absorbance at 600nm of 0.6
| + | ===[https://2013.igem.org/Team:Newcastle/Notebook/protocols#Bacillus_subtilis_168_Competent_cell_Prep_and_Transformation Transformation]=== |
| - | | + | |
| - | • Chill the cells on ice and harvest by centrifugation at 4°C for 10minutes
| + | |
| - | | + | |
| - | • Resuspend the pellet in 100ml of ice cold TFBI
| + | |
| - | | + | |
| - | • Re-centrfuge the cells as before and resuspend in 20ml of ice cold TFBII
| + | |
| - | | + | |
| - | • Aliqot 200µl volumes of the cell suspension into cooled, sterile microfuge tube, an flash freeze in an EtOH/dry ice bath
| + | |
| - | | + | |
| - | • Store the cells at -80°C until requires
| + | |
| - | | + | |
| - | ===Transformation=== | + | |
| - | | + | |
| - | • Thaw a 200µl aliquot of the desired strain of E.coli and add the transforming DNA. Incubate on ice for up to 30-45minutes.
| + | |
| - | | + | |
| - | • Heat-shock the cells in a water bath for 120seconds, and place on ice again for 3-4 minutes
| + | |
| - | | + | |
| - | • Add 1ml of LB broth and incubate the cells at 37°C for 1hour.
| + | |
| - | | + | |
| - | • Spin down the cells in a microfuge (~20seconds) and remove the supernatant
| + | |
| - | | + | |
| - | • Resuspend the cell pellet in ~200µl of broth or d.H2O and plate out on LB (agar at 1.5%), containing the appropriate selection markers.
| + | |
| - | | + | |
| - | • Incubate plates overnight at desired temperature (normally 37°C)
| + | |
| - | | + | |
| - | '''3. Transformed E.coli will be selected through growth on an antibiotic (Chloramphenicol) LB plate.'''
| + | |
| - | | + | |
| - | '''4. Plasmid mini-prep procedure will then be performed in order to purify the plasmids.'''
| + | |
| - | | + | |
| - | '''5. Then the plasmid will be digested with XbaI and SpeI., protocol as follow:'''
| + | |
| - | | + | |
| - | - Using NEBuffer 2.1 at 37oC for 100% activity
| + | |
| - | | + | |
| - | - To make up to 20µl of reaction mix
| + | |
| - | | + | |
| - | - 15µl of DNA/plasmid
| + | |
| - | | + | |
| - | - 1 µl of XbaI and 1 µl SpeI.
| + | |
| - | | + | |
| - | - 2 µl of 10X NEBuffer 2.1 (must be diluted down to 1X)
| + | |
| - | | + | |
| - | - 1 µl of water
| + | |
| - | | + | |
| - | - Incubate the digestion mixture at 37oC for 3 hour
| + | |
| - | | + | |
| - | '''6. 0.7% Agarose gel electrophoresis will then be used in order to separate the DNA fragments.'''
| + | |
| - | | + | |
| - | '''7. Then, perform gel mediated DNA purification in order to purify the ‘switch’ gene construct (2,666bp).'''
| + | |
| - | | + | |
| - | '''8. Prior to transforming this naked DNA (no plasmid), the B. subtilis 168 will be made competent through the following protocol:'''
| + | |
| - | | + | |
| - | ===Materials required for B. subtilis 168 transformation===
| + | |
| - | | + | |
| - | • 10 µl of transformation DNA
| + | |
| - | | + | |
| - | • Pipettes
| + | |
| - | | + | |
| - | • 2X 15 ml falcon tubes
| + | |
| - | | + | |
| - | • 2X 50 ml falcon tubes
| + | |
| - | | + | |
| - | • Eppendorf tubes
| + | |
| - | | + | |
| - | • Agar plates containing the appropriate antibiotic
| + | |
| - | | + | |
| - | • Water bath
| + | |
| - | | + | |
| - | • Centrifuge
| + | |
| - | | + | |
| - | ====SMM medium (1 litre) ====
| + | |
| - | | + | |
| - | 1. 2.0 g of ammonium sulphate
| + | |
| - | | + | |
| - | 2. 14.0 g of dipotassium hydrogen phosphate
| + | |
| - |
| + | |
| - | 3. 6.0 g of potassium dihydrogen phosphate
| + | |
| - | | + | |
| - | 4. 1.0 g of sodium citrate dehydrate
| + | |
| - | | + | |
| - | 5. 0.2 g of magnesium sulphate
| + | |
| - | | + | |
| - | 6. Top up the rest of the medium with water
| + | |
| - | | + | |
| - | ====MM competence medium====
| + | |
| - | | + | |
| - | 1. 10 ml of SMM medium
| + | |
| - | | + | |
| - | 2. 125 µl of Solution E (40% glucose)
| + | |
| - |
| + | |
| - | 3. 100 µl of Tryptophane solution (at a concentration of 2 mg/ml
| + | |
| - | | + | |
| - | 4. 60 µl of Solution F (1M MgSO4)
| + | |
| - | | + | |
| - | 5. 10 µl of 20% Casamino acids
| + | |
| - | | + | |
| - | 6. 5 µl of 0.22% Fe-NH4-citrate
| + | |
| - | | + | |
| - | ====Starvation medium==== | + | |
| - | | + | |
| - | 1. 10 ml of SMM medium
| + | |
| - | | + | |
| - | 2. 125 µl of Solution E (40% glucose)
| + | |
| - | | + | |
| - | 3. 60 µl of Solution F (1M MgSO4)
| + | |
| - | | + | |
| - | ==Protocol==
| + | |
| - | This protocol will stretch for 2 days and aseptic techniques have to be applied for all steps.
| + | |
| - | | + | |
| - | ===Day 1===
| + | |
| - | • Inoculate a single colony of Bacillus subtilis 168 into a 15 ml falcon tube containing 10 ml of MM competence media.
| + | |
| - | | + | |
| - | • For control, transfer 10 ml of MM competence media without the bacteria.
| + | |
| - | | + | |
| - | • Incubate overnight in a shaking incubator at 37oC.
| + | |
| - | | + | |
| - | ===Day 2===
| + | |
| - | • Transfer 0.6 ml of the overnight culture into 50 ml falcon tube containing 10 ml of MM competence medium.
| + | |
| - | • Incubate the tubes for 3 hours at 37oC.
| + | |
| - | | + | |
| - | • Warm up the starvation medium to 37oC in a water bath.
| + | |
| - | | + | |
| - | • Add 10 ml of starvation medium (prewarmed) into each tube and incubate for a further 1-2 hours at 37oC.
| + | |
| - | | + | |
| - | • Transfer 0.4 ml of the medium not containing B. subtilis into one Eppendorf tube.
| + | |
| - |
| + | |
| - | • Add 10 µl of water into the tube.
| + | |
| - | | + | |
| - | • Transfer 0.4 ml of the medium containing B. subtilis into two Eppendorf tubes.
| + | |
| - | | + | |
| - | • Add 10 µl of DNA into one tube.
| + | |
| - | | + | |
| - | • Add 10 µl of water into one tube.
| + | |
| - | | + | |
| - | • Incubate the samples for 1 hour at 37oC in the shaking incubator.
| + | |
| - | | + | |
| - | • The Eppendorf tubes have to be aerated, therefore incubate the tubes on their side.
| + | |
| - | | + | |
| - | • Centrifuge the Eppendorf tubes at 13,000 rpm for 2 minutes.
| + | |
| - | | + | |
| - | • Discard 0.3 ml of supernatant from each Eppendorf tubes.
| + | |
| - | | + | |
| - | • Resuspend the pellet in the remaining supernatant and plate it onto agar plates containing the appropriate antibiotic.
| + | |
| - | • Incubate the plates overnight at 37oC.
| + | |
| - | | + | |
| - | '''9. These competent cells will then be transformed with the naked DNA. The homologous region on the 5’ (pbpB) and 3’ (murE) will cause homologous recombination event. This Double cross over even will swap its native spoVD gene with the BioBrick.'''
| + | |
| | | | |
| | [[File:L-form biobrick construct.png|500px]] | | [[File:L-form biobrick construct.png|500px]] |
| Line 220: |
Line 74: |
| | There are 2 main approaches to do this: | | There are 2 main approaches to do this: |
| | | | |
| - | ====Liquid media==== | + | ==== [https://2013.igem.org/Team:Newcastle/Notebook/protocols#L-form_Growth_Media Liquid media]==== |
| - | | + | |
| - | • Mix equal volumes of 2xMSM and 2xNutrient Broth (Oxoid,NB) and add 5µg/ml of Chloramphenicol
| + | |
| - | | + | |
| - | • Add14-20mL of media to a 250ml Erlenmeyer flask/ 8-10 mL into 100 ml flask (to allow a large interface surface with air that will ensure proper oxygenation0
| + | |
| - | | + | |
| - | • From the Xylose supplemented media, pick a few colonies and inoculate the media
| + | |
| - | | + | |
| - | • Gently mix the media to break up the cells
| + | |
| - | | + | |
| - | • Incubate at 30oC without shaking
| + | |
| - | | + | |
| - | • Leave to grow for 1-2 days
| + | |
| - | | + | |
| - | Notes: A large media volume to flask volume ratio allows a thin layer of media which ensures diffusion of oxygen. Shaking is not necessary for growth, and if done should be very gently.
| + | |
| - | | + | |
| - | ====Lysozyme protoplasting method==== | + | |
| - | • Take the cells from Xylose supplemented plate and put it in 4mL of LB+xyl (0.8%) and incubate at 37oC for 2-3 hours
| + | |
| - | | + | |
| - | • Dilute the cells from the 4mL media into the 10ml LB + 0.8% xylose media
| + | |
| - | | + | |
| - | • Check OD of cells every 1 hour and make sure it goes between 0.3-0.6 (mid exponential stage)
| + | |
| - | | + | |
| - | • Spin down @~5,500rpm for 4 minutes to remove the xylose
| + | |
| - | | + | |
| - | • Wash once with LB
| + | |
| - | | + | |
| - | • Pellet the cells @4,500rpm for 15 minutes
| + | |
| - | | + | |
| - | • Resuspend pellet with Lysis solution (NB/MSM (2-4mL) + PenG (200µg/ml) + Lysozyme (2-4 mg/ml))
| + | |
| - | | + | |
| - | • Incubate for 1-1.30 hours @ 37oC for the lysozyme to work
| + | |
| - | | + | |
| - | • Check under microscope between the incubation period until 99.99% of cells are protoplast
| + | |
| - | | + | |
| - | • Spin down and re-suspend with NB/MSM (10-15mL) + Chloramphenicol (5µg/ml) + 200µl of 200mg/ml PenG
| + | |
| | | | |
| - | • Incubate @ 30oC for at least 2 days
| + | ==== [https://2013.igem.org/Team:Newcastle/Notebook/protocols#Protoplasting_to_generate_L-form Lysozyme protoplasting method]==== |
| | | | |
| | {{Team:Newcastle/Sponsors}} | | {{Team:Newcastle/Sponsors}} |
Switch BioBrick
Purpose and justification
The purpose of this main ‘keystone’ biobrick is to facilitate the switching of Bacillus subtilis cells from rod-shape to L-form, wall-less cells. Furthermore, this ‘switch’ should also enable L-form cells to return to rod-shape when required.
This will be facilitated through the introduction of a xylose-controlled promoter (PxylR) upstream of the murE gene, which is involved in the biosynthesis of peptidoglycan. The presence, or absence of a cell wall would be controlled through the presence, or absence of xylose, respectively.
The antibiotic resistance marker chloramphenicol acetyl-transferase (cat) is upstream of this promoter region to allow for the selection of cells, in which this system is integrated.
Homologous recombination will allow this biobrick to be integrated into the chromosome of B. subtilis. The cat and PxylR sequences will be flanked by sequences that are homologous with regions of the B. subtilis chromosome, which will allow insertion at the intended loci. ~300bp at the 5’ end of the biobrick will be homologous with the end of the pbpB gene. Similarly ~300bp at the 3’ end of the biobrick will be homologous with the start of the murE gene.
Modelling
Modelling biosynthesis of peptidoglycan, specifically involving manipulation of the murE gene.
Currently undergoing completion…
Construction
To be synthesised by DNA 2.0.
Parts:
1. Biobrick prefix – RFC 10 standard.
2. 309bp of the end of the pbpB coding sequence.
3. Spacer.
4. Reverse and complement of chloramphenicol acetyl-transferase (cat) coding sequence (including native ribosome binding site (RBS) and promoter).
5. PxylR promoter.
6. Spacer.
7. RBS binding site for murE.
8. Spacer.
9. 331bp of the start of murE coding sequence.
10. Biobrick suffix – RFC 10 standard.
Restriction map of biobrick (without biobrick prefix and suffix):

Produced using Clone Manager 9 Professional Edition
Cut sites include PstI and EcoRI (x2) – these are in the RFC 10 prefix and suffix restriction sites. The sequence will need to be altered to prevent being cut during excision of the biobrick.
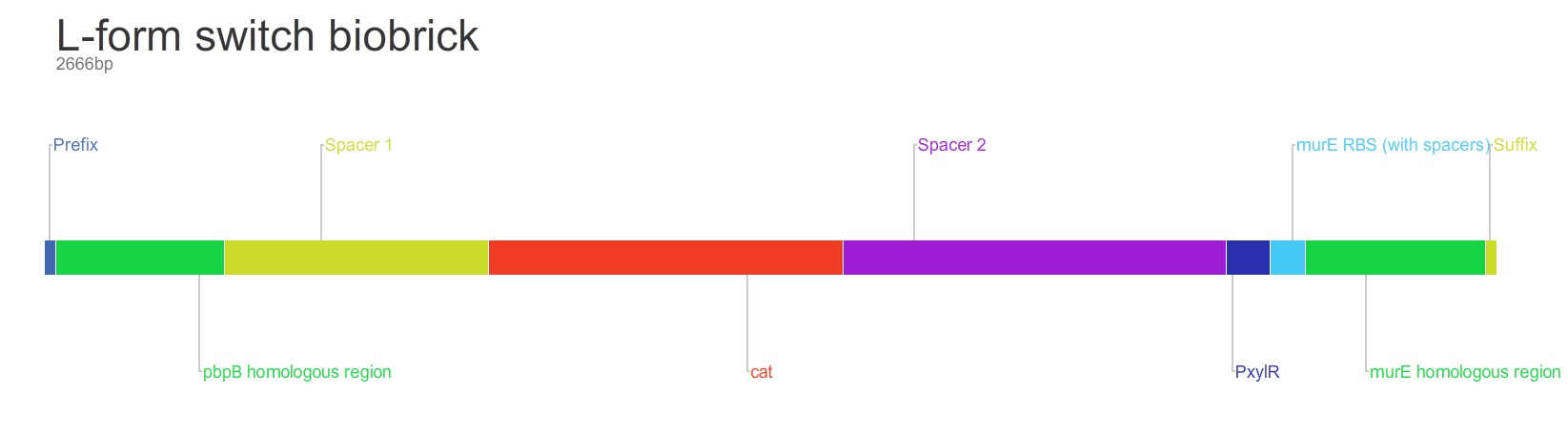
Produced using Gene Designer 2.0
Cloning and integration
1. The gene construct will be clone into pSB1C3, which is the only plasmid acceptable by iGEM for part submission.
2. The plasmid that is returned from DNA2.0 will be amplified in E.coli. The following protocol outlines the procedure by which this will take place:

Testing and Characterisation
Transformants will be selected through growth on LB+Chloramphenicol+0.5-0.8% xylose media. Only those which took up the DNA and integrate the DNA will survive.
To characterise this BioBrick, the B. subtilis is to be grown on a media without xylose and then the morphology of the cells will be checked under the microscope. Successfully transformed B. subtilis cells should have converted to L-form state (lost their cell walls). These are recognisable due to their rounded and non-uniform morphology. Cells may also vary greatly in size.
There are 2 main approaches to do this:
 "
"



