 "
"
Team:UGent/Results
From 2013.igem.org
| (56 intermediate revisions not shown) | |||
| Line 2: | Line 2: | ||
{{:Team:UGent/Templates/Navigation}} | {{:Team:UGent/Templates/Navigation}} | ||
<html> | <html> | ||
| + | <h1> Strains with <i>ccdA-gfp</i> construct </h1> | ||
| + | <p> | ||
| + | The following strain containing the construct to be duplicated (<i>ccdA-gfp</i>) was constructed (KI experiment 1): | ||
| + | <center><b><i>E. coli</i> MG1655 ΔendA DE3 ccdA-Pmb1GFP-CmFRT</b></center> | ||
| + | </p> | ||
| + | <table> | ||
| + | <tr> | ||
| + | <td> | ||
| + | <ul> | ||
| + | <li> Colony PCR on 48 colonies using crimson taq polymerase with two primer pairs: MDM0141/MDM0010 and MDM0046/MDM123. Expected fragments: 550bp & 2450 bp. Analytic gel: <b> 4 positives: colonies 26, 28, 35 and 36 </b> | ||
| + | </li> | ||
| + | </ul> | ||
| + | </td> | ||
| + | <td> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/0/02/UGent_2013_CPCR_KI.jpg" width="150"/></a> | ||
| + | </td> | ||
| + | </tr> | ||
| + | <tr> | ||
| + | <td> | ||
| + | <ul> | ||
| + | <li> Colony PCR on 4 positive and 4 negative colonies using Emerald polymerase with out primers: MDM0046/MDM0010. Expected fragment: ca. 5500 bp. Analytic gel: <b> 1 positive: colonie 35 </b>.</li></ul> | ||
| + | </td> | ||
| + | <td> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/3/35/UGent_2013_CPCR_KI_out.png" width="150"/></a></li> | ||
| + | </td> | ||
| + | </tr> | ||
| + | <tr> | ||
| + | <td> | ||
| + | <ul> | ||
| + | <li> After inoculation of c35 on cm plate: Colony PCR on 8 kolonies using emerald polymerase with out primers: MDM0046/MDM0010. Expected fragment: ca. 5500 bp. Analytic gel: <b> positive: 5 & 8 </b>.</li> | ||
| + | </ul> | ||
| + | </td><td> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/8/85/CPCR_KI_out2.png" width="150"><td> | ||
| + | </tr> | ||
| + | </table> | ||
| + | <p>We have two versions of this strain: 5 and 8. The two strains were used in our further experiments as to test possible differences between the two versions. | ||
| + | </p> | ||
| + | |||
<h1> Plasmids containing T7-<i>ccdB</i> </h1> | <h1> Plasmids containing T7-<i>ccdB</i> </h1> | ||
<p> | <p> | ||
| - | We constructed plasmid pSB6A1-T7<i>ccdB</i> for use in CIChE. Through | + | We constructed plasmid pSB6A1-T7<i>ccdB</i> for use in CIChE and pSB1C3-T7<i>ccdB</i> to submit in the biobrick-registry. Through these plasmids, pressure can be put on the CIChE strains simply by adding IPTG. We started with a PCR on p20SpFRT-T7<i>ccdB</i> with primers MDM0586 and MDM0587. We then used restriction enzymes XbaI and PstI to cut the obtained T7-<i>ccdB</i> and the plasmid backbones (pSB6A1 and pSB1C3). After ligating these parts, the DNA was transformed into <i>E. coli</i> Top10 subcloning cells, using heat shock. This experiment delivered pSB6A1 with a deletion of cytosine at position 5929 (for pSB6A1), but failed multiple times for pSB1C3. We then tried to create the plasmid using the Gibson Assembly technique. This was done using T7-<i>ccdB</i> and backbone constructed with specific primers. The result of this transformation (using electroporation) was pSB1C3 with a deletion of adenosine at position 3988 (for pSB1C3). A backmutation was performed on both plasmids using specially constructed primers. This process was <b>succesfull for pSB6A1-T7<i>ccdB</i></b>, but not for pSB1C3-T7<i>ccdB</i>. |
| + | |||
| + | |||
| + | |||
<br><br> | <br><br> | ||
| - | We also purified plasmids p5SpFRT-T7<i>ccdB</i>, p10SpFRT-T7<i>ccdB</i>, p20SpFRT-T7<i>ccdB</i> from strains we obtained from | + | We also purified plasmids p5SpFRT-T7<i>ccdB</i>, p10SpFRT-T7<i>ccdB</i>, p20SpFRT-T7<i>ccdB</i> from strains we obtained from InBio. These plasmids have different copy numbers and they too can be used in chromosomal evolution. |
<br><br> | <br><br> | ||
Having plasmids with different copy numbers at our disposal, we can test CIChE with different degrees of toxin-pressure. | Having plasmids with different copy numbers at our disposal, we can test CIChE with different degrees of toxin-pressure. | ||
</p> | </p> | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
<h1> CIChE Strains</h1> | <h1> CIChE Strains</h1> | ||
<p> | <p> | ||
The plasmids and strains mentioned above were used in transformations (experiment 3) to obtain strains containing all elements necessary to perform chromosomal evolution. The following strains were constructed:<br> | The plasmids and strains mentioned above were used in transformations (experiment 3) to obtain strains containing all elements necessary to perform chromosomal evolution. The following strains were constructed:<br> | ||
| - | <table | + | <table> |
<tr> | <tr> | ||
| - | <td> | + | <td width="270"> |
| + | |||
<ul> | <ul> | ||
<li>8+pSB6A1-T7<i>ccdB</i></li> | <li>8+pSB6A1-T7<i>ccdB</i></li> | ||
| + | </ul> | ||
| + | </td> | ||
| + | <td> | ||
| + | <b>Colony PCR</b> on 8 + pSB6A1 using emerald polymerase with primerpairs MDM0046/MDM010 to check KI. Expected fragment: 5500bp. Analytic gel: <b> positive </b> | ||
| + | <br><br> | ||
| + | <b>Colony PCR</b> on 8 + pSB6A1 using Q5 polymerase to test for pSB6A1 <b>after backmutation</b> with primers MDM0606 and MDM0607). Expected fragment: 4387 bp. Analytic gel: <b> positive </b> | ||
| + | </td> | ||
| + | <td> | ||
| + | <table> | ||
| + | <tr> | ||
| + | <td> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/0/08/UGent_2013_Control_ccda_competente_cellen.jpg" width="50" height="205"/> | ||
| + | </td> | ||
| + | <td> | ||
| + | <img src=" https://static.igem.org/mediawiki/2013/9/9d/UGent_2013_pSB6A1_cPCR_w3_sept.png " width="150"/> | ||
| + | </td> | ||
| + | </tr> | ||
| + | </table> | ||
| + | </td> | ||
| + | </tr> | ||
| + | <td> | ||
| + | <ul> | ||
<li>8+p5SpFRT-T7<i>ccdB</i></li> | <li>8+p5SpFRT-T7<i>ccdB</i></li> | ||
<li>8+p10SpFRT-T7<i>ccdB</i></li> | <li>8+p10SpFRT-T7<i>ccdB</i></li> | ||
<li>8+p20SpFRT-T7<i>ccdB</i></li> | <li>8+p20SpFRT-T7<i>ccdB</i></li> | ||
| + | <br><br> | ||
| + | <li>5+p5SpFRT-T7<i>ccdB</i></li> | ||
| + | <li>5+p10SpFRT-T7<i>ccdB</i></li> | ||
| + | <li>5+p20SpFRT-T7<i>ccdB</i></li> | ||
</ul> | </ul> | ||
</td> | </td> | ||
<td> | <td> | ||
| - | < | + | <b>Colony PCR</b> on 5 and 8 + pSB6A1/p5/p10/p20 using emerald polymerase with primerpairs MDM0046/MDM010 to check KI. Expected fragment: 5500bp. Analytic gel: <b> positive </b> |
| - | < | + | <br><br> |
| - | < | + | <b>Colony PCR</b> on 5 and 8 + pSB6A1/p5/p10/p20 using emerald polymerase with primerpairs MDM0096/CGL0019 for pSB6A1 and MDM0039/MDM0060 for p5/p10/p20. Expected fragment: 437bp & 910 bp. Analytic gel: <b> positive for p5/p10/p20</b> |
| - | < | + | </td> |
| - | </ | + | <td> |
| + | <img src="https://static.igem.org/mediawiki/2013/8/88/UGent_2013_CPCR_MDMcm.jpg" width="150"><br> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/e/e0/UGent_2013_CPCR_check_plasmids_strain_5.jpg" width="150"> | ||
</td> | </td> | ||
</tr> | </tr> | ||
</table> | </table> | ||
| + | |||
| + | |||
</p> | </p> | ||
<h1> UV Test </h1> | <h1> UV Test </h1> | ||
| - | <p> Phage transduction was used to perform the deletion of the <i>recA</i> gene after chromosomal evolution. To check whether the gene was successfully knocked out, | + | <p> Phage transduction was used to perform the deletion of the <i>recA</i> gene after chromosomal evolution. To check whether the gene was successfully knocked out, a UV test was developed. The UV light puts a certain amount of stress on the bacterial cells. In respons to this stress, an SOS pathway is triggered in which <i>recA</i> plays an important role. Cells in which the <i>recA</i> gene is deleted will not survive on the UV-treated plate. Colonies in which <i>recA</i> was successfully deleted still can be found on a non-treated backup plate. |
<br><br> | <br><br> | ||
<b>RESULT: </b> | <b>RESULT: </b> | ||
| Line 72: | Line 141: | ||
<h1> CIChE </h1> | <h1> CIChE </h1> | ||
<p> | <p> | ||
| - | + | The practical execution of CIChE was as follows: a strain was inoculated with the right concentration of IPTG (day 1 = D1), these strains were grown overnight at 37°C. The next day the strains were inoculated with the same IPTG concentration (day 2 = D2) and with an increased IPTG level (D1). IPTG concentration was raised with a small “step” or with a larger “jump”. These strains were also grown overnight at 37°C. This was repeated over the next days. Because the outcome was difficult to predict, a lot of different IPTG concentrations and ranges were tested. | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
<br><br> | <br><br> | ||
| - | + | To test all of our CIChE strains we used 22 microtiter plates (the distribution of the different strains in the plates is documented in <a href="https://static.igem.org/mediawiki/2013/2/22/UGent_2013_Plates_complete.pdf" target="_blank">this pdf file</a>). To spare some time we used the second, faster protocol (see <a href="https://2013.igem.org/Team:UGent/Experiments" target="_blank">experiment 5</a>). Bacteria were grown in microtiter plates during 4 hours at 37°C on shakers. Afterwards the OD (optical density) and fluorescence intensity (F) were measured inside the FLUOstar. Strains with more copies of the <i>gfp</i> gene should result in higher fluorescence intensity, indicating a higher construct copy number. Because the fluorescence not only depends on the gene copy number, but also on the OD, we divided the fluorescence by the OD (F/OD) to analyse our results. | |
| - | < | + | <br><br> |
| + | The plates also contained blank wells and reference colonies. These were necessary for two different reasons. | ||
| + | </p><ul> | ||
| + | <li>We subtracted the OD of all the wells with the average OD of the blank and did the same for the fluorescence intensity. This was done to standardize the measurements. </li> | ||
| + | <li>Because we used many different plates (and thus some strains were spread over two or more plates), we added a reference strain (strain 8) to all the plates. This strain contains one copy of our construct and thus should give the same F/OD in every well. We used various statistical tests to check if there was a significant difference in average F/OD between the plates of one strain. If these tests resulted in a significant difference of the 8 strain, we could not compare the measurements. Luckily this only happened for a few strains. The extensive execution of these tests can be checked in <a href="https://static.igem.org/mediawiki/2013/d/db/UGent_2013_statistics.pdf" target="_blank">this pdf file</a>.</li></ul><p> | ||
| + | Thereafter we calculated the F/OD for every well. If the data showed some abnormalities, we tested them for outliers. If the values of the outliers were significantly different than the values of the rest of the data, we excluded these observations and did not use them in our further analysis. | ||
| + | <br><br> | ||
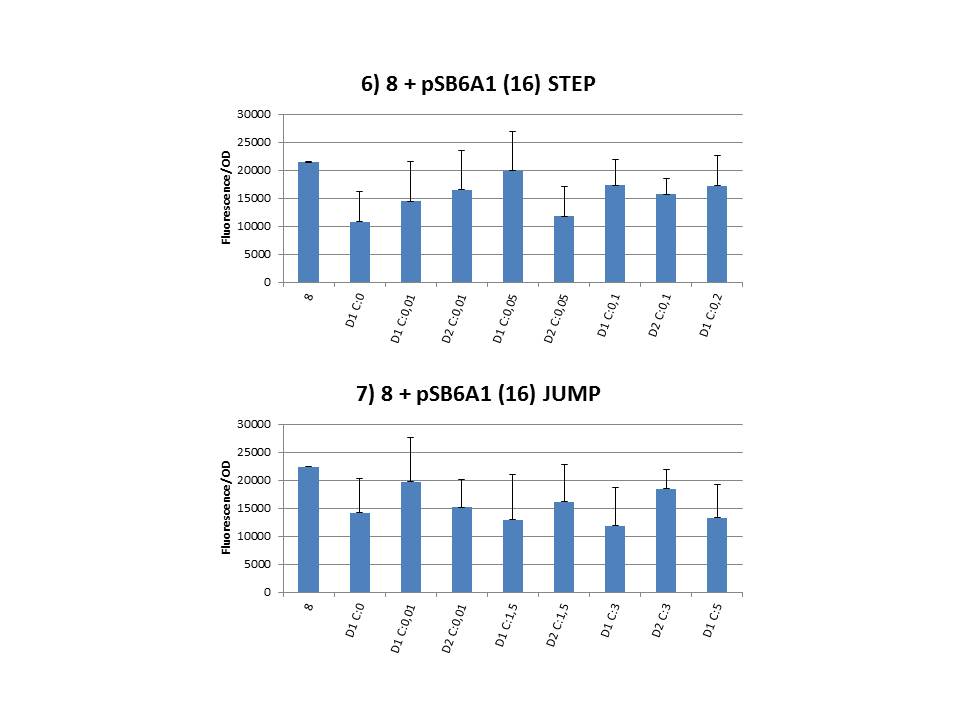
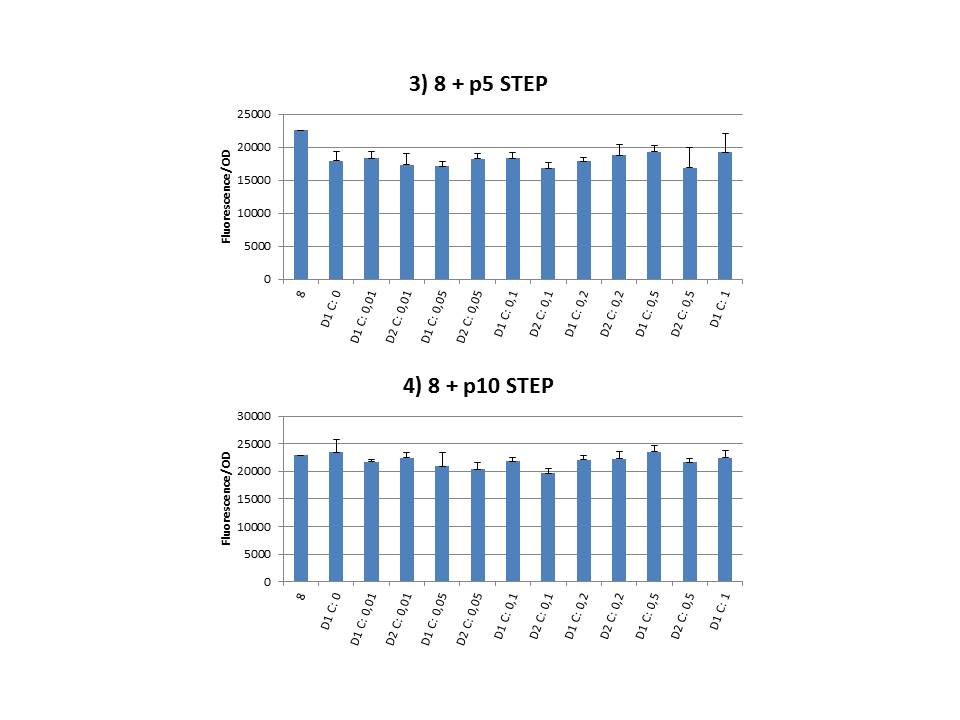
| + | Finally we plotted the F/OD in function of the IPTG concentration. The results can be seen in figure 1-9 (you can click on the graphs to enlarge). | ||
| + | <br><br> | ||
| + | In every graph the average F/OD of strain 8 is added as reference. The average is calculated using the results of 8 corresponding to the correct plates. If our model for CIChE works, our strains (containing more gfp copies) should have an average F/OD higher than the one of strain 8. We also expected that the F/OD would rise with increasing IPTG concentrations. Looking at the graphs we see that most of our strains result in an average F/OD lower than or about the same as the average result of strain 8. This is not at all what we expected/hoped. <b>According to the obtained data, we must conclude that no duplication of the construct occurred, hence no chromosomal evolution was achieved. However, we have a pretty good idea what lead to these results. Our hypothesis can be read below.</b> | ||
</p> | </p> | ||
| - | < | + | <table> |
| - | + | <td><a href="https://static.igem.org/mediawiki/2013/f/f3/UGent_2013_plot1.JPG" target="_blank"><img src="https://static.igem.org/mediawiki/2013/f/f3/UGent_2013_plot1.JPG" width="400"/></a></td> | |
| - | <br> | + | <td><a href="https://static.igem.org/mediawiki/2013/9/9f/UGent_2013_plot2.JPG" target="_blank"><img src="https://static.igem.org/mediawiki/2013/9/9f/UGent_2013_plot2.JPG" width="400"/></a></td> |
| - | <br> | + | <tr> |
| + | <td><a href="https://static.igem.org/mediawiki/2013/3/35/UGent_2013_plot3.JPG" target="_blank"><img src="https://static.igem.org/mediawiki/2013/3/35/UGent_2013_plot3.JPG" width="400"/></a></td> | ||
| + | <td><a href="https://static.igem.org/mediawiki/2013/1/19/UGent_2013_plot4.JPG" target="_blank"><img src="https://static.igem.org/mediawiki/2013/1/19/UGent_2013_plot4.JPG" width="400"/></a></td> | ||
| + | </tr> | ||
| + | <tr> | ||
| + | <td><a href="https://static.igem.org/mediawiki/2013/c/c5/UGent_2013_plot5.JPG" target="_blank"><img src="https://static.igem.org/mediawiki/2013/c/c5/UGent_2013_plot5.JPG" width="400"/></a></td> | ||
| + | </tr> | ||
| + | </table><br> | ||
| + | <h1>CIChE and its stumbling blocks: our hypothesis</h1> | ||
| + | <p>For the construction of our plasmid we chose to work with a T7 promoter for the <i>ccdB</i> gene as to avoid leaky expression. Cloning was performed in Top10 cells which do not contain T7 RNA polymerase. Taking into account the troubles we had with the construction of the plasmids with the T7-<i>ccdB</i> insert, we can almost certainly conclude that despite this the cells did experience hindering of the CcdB toxin. Often we obtained no colonies after transformation and if we did, there were mutations in the toxic part of the <i>ccdB</i> gene (see further). We presume that this can only be caused if the <i>ccdB</i> was transcribed by other polymerases who are not specific for the T7 promoter. This probably only occurs at very low frequencies, but as in these strains there is no CcdA present at all even the lowest amount of working CcdB can be lethal for these cells.<br><br> | ||
| + | We performed CIChE with the plasmid we constructed ourselves (pSB6A1-T7<i>ccdB</i>) as well as with the original plasmids from which we constructed this part: p5SpFRT-T7<i>ccdB</i>, p10SpFRT-T7<i>ccdB</i> and p20SpFRT-T7-<i>ccdB</i>. According to the GFP measurements we performed, none of these resulted in duplication of the <i>ccdA-gfp</i> construct in the genome. We presume that this is caused by the malfunction of CcdB due to mutations. But how is it possible that there’s so many of these mutations?<br><br> | ||
| + | It is known from previous research that the part essential for CcdB’s toxic activity is situated at the last three amino acids (Trp-Gly-Ile). Interaction between CcdB and the DNA gyrase A subunit (GyrA) takes place at Trp99 from CcdB and Arg462 of GyrA59 (the 59 kDa amino-terminal breaking-rejoining domain of GyrA). Random mutations in the final part of the <i>ccdB</i> gene affecting this toxic site will consequently provide these cells with a great growth advantage over cells with intact <i>ccdB</i> genes. We assume this is a possible reason why mutation frequency of the <i>ccdB </i>gene seems to be so high in our project.<br><br> | ||
| + | Furthermore, in the absence of its antidote CcdA, CcdB causes reduced DNA synthesis, activation of the SOS pathway, cell filamentation and eventually cell death. One of the aspects of this SOS pathway is the enhancing of capacity of mutagenesis, such as point and frameshift mutations. So if for example there is leaky expression of the promoter and CcdB is being formed without a sufficient amount of CcdA present, SOS pathway will be activated causing higher mutation frequencies. We postulate that these mutations can also affect the ccdB gene itself and thus restoring ‘normal’ cell life. In combination with what we discussed in the previous paragraph this could explain the amount of mutations we had to deal with over the course of our project.<br><br> | ||
| + | We chose some strains at random during the CIChE experiment (8 + pSB6A1-T7<i>ccdB</i>, 2 mM IPTG day 2, Step) and let the plasmid get sequenced. When we compared the ccdB sequence to the native one we saw our plasmid had a deletion of a cytosine residue at position 297 (out of a total of 306 base pairs). <b>This causes a frameshift, resulting in a prolonged protein</b> of 154 amino acids instead of the native structure with 102 amino acids. <i>(Note: CcdB normally contains 101 amino acids, but the sequence we used has an extra Alanine after the start codon for cloning purposes.)</i> Likewise, all pSB1C3-T7<i>ccdB </i>plasmids we sent for sequencing contained a mutation at the end: all of them had a deletion of the A residu of the stop codon, also leading to a frameshift and prolonging of the protein.<br><br> | ||
| + | As mentioned before, the toxic part of CcdB is situated at the last three amino acids. None of the sequencing results showed a mutation anywhere in the beginning or middle of the <i>ccdB </i>gene. <b>This shows the mutations are really all linked with the toxicity and cells lacking toxic CcdB easily overrule the cells that do contain toxic CcdB. If experimental conditions are not adapted there’s little chance of success.</b> | ||
| + | <br></p> | ||
| + | <h2>Clone Manager and I-TASSER</h2> | ||
| + | <p>In order to examine the effect of these mutations on the protein structure, we determined the amino acid sequence of the mutated plasmid 8 + pSB6A1-T7<i>ccdB</i>, 2 mM IPTG day 2, Step using Clone Manager:<br></p><p> | ||
| + | MAQFKVYTYKRESRYRLFVDVQSDIIDTPGRRMVIPLASARLLSDKVSRELYPVVHIGDESWRMMTTDMASVPVSVI<br> | ||
| + | GEEVADLSHRENDIKNAINLMFGEYNLGEVKRRSADGSVGSPHARVGNCQASNKTKGSVERLGLSFYLLFVGERSPE<br></p><p> | ||
| + | At the position of the Tryptophan residue responsible for interaction with Arg462 and consequently essential for toxic activity, there is now a Glycine residue. Also the following two amino acids essential for toxicity are mutated. Furthermore chances are that the following part of the protein will sterically hinder interaction with DNA gyrase. <b>As a conclusion this amino acid sequence is a good indication that this mutated CcdB protein won’t be able to execute its toxic function.</b> | ||
| + | To further investigate the consequences of the frameshift we used I-TASSER to predict the structure of this protein, resulting in following top 5 of predicted models:<br></p> | ||
| + | <table> | ||
| + | <tr> | ||
| + | <td> | ||
| + | <img src=" https://static.igem.org/mediawiki/2013/0/0f/UGent_2013_Model1.gif" width="180" > | ||
| + | </td> | ||
| + | <td> | ||
| + | <img src=" https://static.igem.org/mediawiki/2013/6/6e/UGent_2013_Model2.gif" width="180" > | ||
| + | </td> | ||
| + | <td> | ||
| + | <img src=" https://static.igem.org/mediawiki/2013/d/d5/UGent_2013_Model3.gif" width="180" > | ||
| + | </td> | ||
| + | <td> | ||
| + | <img src=" https://static.igem.org/mediawiki/2013/4/48/UGent_2013_Model4.gif " width="180" > | ||
| + | </td> | ||
| + | <td> | ||
| + | <img src=" https://static.igem.org/mediawiki/2013/f/f7/UGent_2013_Model5.gif" width="180" > | ||
| + | </td> | ||
| + | </tr> | ||
| + | <tr> | ||
| + | <td> | ||
| + | <center>C-score = -2.27 </center> | ||
| + | </td> | ||
| + | <td> | ||
| + | <center>C-score = -2.76</center> | ||
| + | </td> | ||
| + | <td> | ||
| + | <center>C-score = -2.75</center> | ||
| + | </td> | ||
| + | <td> | ||
| + | <center>C-score = -2.69</center> | ||
| + | </td> | ||
| + | <td> | ||
| + | <center>C-score = -2.93</center> | ||
| + | </td> | ||
| + | </tr> | ||
| + | <tr> | ||
| + | <td> | ||
| + | </table> | ||
| + | <p><i>Note: C-score is typically in the range of [-5,2], where a C-score of higher value signifies a model with a high confidence and vice-versa.</i><br><br> | ||
| + | Whereas native CcdB has an α helix at its C-terminal end, two of these end with a β strand and three with an undetermined structure.<br><br> | ||
| + | Five of the top 10 templates used by I-TASSER are CcdB from E. coli and four of them are CcdB from <i>V. fischeri</i>. This shows that to a great extent there still is resemblance to the native <i>ccdB</i> sequence. The tenth template used by I-TASSER is the A subunit of DNA gyrase, the target of CcdB. | ||
| + | When comparing the proposed structure of our mutated plasmid to the native CcdB structure (pdb entry 3VUB) using I-TASSER this is what we get, with <b>in red the native CcdB structure and in blue our mutated protein</b>. The red, native, part overlaps practically completely with the blue one. The rest of the blue structure is the extension caused by the frameshift. <br><br> | ||
| + | |||
| + | |||
| + | <center><img src="https://static.igem.org/mediawiki/2013/4/4a/UGent_2013_Model1_3vubA.gif" width="250"></center><br> | ||
</p> | </p> | ||
| + | |||
| + | |||
<h1>Further research perspectives </h1> | <h1>Further research perspectives </h1> | ||
<p> | <p> | ||
| - | <b> Assemble T7-ccdB plasmid inside CcdB-survival strains </b | + | <b> Assemble T7-<i>ccdB</i> plasmid inside CcdB-survival strains </b><br> |
| - | When assembling the plasmid containing T7-ccdB in cells resistant to CcdB, mutations as a result of stress are less likely to occur at this stage. We experienced difficulties cloning the T7-ccdB construct in our plasmids probably because of leaky expression of the T7-promoter. | + | When assembling the plasmid containing T7-<i>ccdB</i> in cells resistant to CcdB, mutations as a result of stress are less likely to occur at this stage. We experienced difficulties cloning the T7-<i>ccdB</i> construct in our plasmids probably because of leaky expression of the T7-promoter. |
<br><br> | <br><br> | ||
| - | <b> | + | <b>Cloning T7-<i>ccdB</i> plasmid in <i>ccdA</i>-strain</b><br> |
| + | If the plasmids are cloned in the strains that already contain the <i>ccdA</i> construct, leaky expression of <i>ccdB</i> might be neutralized by the CcdA already present. This would lower the toxin pressure, making mutations less likely to occur. | ||
| + | <br><br> | ||
| + | <b>Adding multiple <i>ccdA</i> copies to the <i>ccdA-gfp</i> construct</b><br> | ||
| + | If multiple <i>ccdA</i> copies were incorporated in the CIChE-construct, cells would be able to handle higher amounts of CcdB toxin. They would not feel pressured as much to mutate. | ||
| + | <br><br> | ||
| + | <b>Knock out of alternative polymerases</b><br> | ||
| + | <i>E. coli</i> possesses five DNA polymerases. Polymerase I and III see to respectively the maturation of Okazaki fragments and replication under vegetative conditions. Polymerase II, IV and V are the so called alternative polymerases or error prone DNA polymerases. They can be induced under a variety of environmental stress factors and mediate stress-induced mutagenesis. They also mediate translesion synthesis and enable efficient replication past DNA damage that would otherwise halt replication. Unfortunately this happens with significantly reduced fidelity, resulting in a higher mutation rate. A possible solution to reduce this rate could be the knock out (and maybe the knock in of high fidelity polymerases) of these low fidelity polymerases.<br><br> | ||
| + | <b>Other alternatives</b><br> | ||
| + | In our literature study we discussed some more alternatives, you can read about them <a href="https://static.igem.org/mediawiki/2013/d/dc/UGent_2013_Bachelorproef.pdf" target="_blank">here</a> (page 6-20). | ||
| + | |||
| + | </p> | ||
| + | <br><br> | ||
| + | <h1>Sources</h1> | ||
| + | Dao-Thi, M.H. et al. Molecular basis of gyrase poisoning by the addiction toxin CcdB. <i>Journal of Molecular Biology</i> <b>348</b>, 1091-1102 (2005). | ||
| + | <br>Couturier, M., Bahassi, E. & Van Melderen, L. Bacterial death by DNA gyrase poisoning. <i>Trends in Microbiology</i> <b>6</b>, 269-275 (1998). | ||
| + | <br>Corzett, C.H., Goodman, M.F. & Finkel, S.E. Competitive Fitness During Feast and Famine: How SOS DNA Polymerases Influence Physiology and Evolution in <i>Escherichia coli</i>. <i>Genetics</i> <b>194</b>, 409-420 (2013). | ||
| - | |||
</html> | </html> | ||
{{:Team:UGent/Templates/Footer}} | {{:Team:UGent/Templates/Footer}} | ||
{{:Team:UGent/Templates/BaseSponsor}} | {{:Team:UGent/Templates/BaseSponsor}} | ||
Latest revision as of 11:34, 15 October 2013
 |
||||||||||||||||||||||||||||||||||
|
Strains with ccdA-gfp constructThe following strain containing the construct to be duplicated (ccdA-gfp) was constructed (KI experiment 1):
We have two versions of this strain: 5 and 8. The two strains were used in our further experiments as to test possible differences between the two versions. Plasmids containing T7-ccdB
We constructed plasmid pSB6A1-T7ccdB for use in CIChE and pSB1C3-T7ccdB to submit in the biobrick-registry. Through these plasmids, pressure can be put on the CIChE strains simply by adding IPTG. We started with a PCR on p20SpFRT-T7ccdB with primers MDM0586 and MDM0587. We then used restriction enzymes XbaI and PstI to cut the obtained T7-ccdB and the plasmid backbones (pSB6A1 and pSB1C3). After ligating these parts, the DNA was transformed into E. coli Top10 subcloning cells, using heat shock. This experiment delivered pSB6A1 with a deletion of cytosine at position 5929 (for pSB6A1), but failed multiple times for pSB1C3. We then tried to create the plasmid using the Gibson Assembly technique. This was done using T7-ccdB and backbone constructed with specific primers. The result of this transformation (using electroporation) was pSB1C3 with a deletion of adenosine at position 3988 (for pSB1C3). A backmutation was performed on both plasmids using specially constructed primers. This process was succesfull for pSB6A1-T7ccdB, but not for pSB1C3-T7ccdB.
CIChE Strains
The plasmids and strains mentioned above were used in transformations (experiment 3) to obtain strains containing all elements necessary to perform chromosomal evolution. The following strains were constructed:
UV Test Phage transduction was used to perform the deletion of the recA gene after chromosomal evolution. To check whether the gene was successfully knocked out, a UV test was developed. The UV light puts a certain amount of stress on the bacterial cells. In respons to this stress, an SOS pathway is triggered in which recA plays an important role. Cells in which the recA gene is deleted will not survive on the UV-treated plate. Colonies in which recA was successfully deleted still can be found on a non-treated backup plate.
CIChE
The practical execution of CIChE was as follows: a strain was inoculated with the right concentration of IPTG (day 1 = D1), these strains were grown overnight at 37°C. The next day the strains were inoculated with the same IPTG concentration (day 2 = D2) and with an increased IPTG level (D1). IPTG concentration was raised with a small “step” or with a larger “jump”. These strains were also grown overnight at 37°C. This was repeated over the next days. Because the outcome was difficult to predict, a lot of different IPTG concentrations and ranges were tested.
Thereafter we calculated the F/OD for every well. If the data showed some abnormalities, we tested them for outliers. If the values of the outliers were significantly different than the values of the rest of the data, we excluded these observations and did not use them in our further analysis.
CIChE and its stumbling blocks: our hypothesisFor the construction of our plasmid we chose to work with a T7 promoter for the ccdB gene as to avoid leaky expression. Cloning was performed in Top10 cells which do not contain T7 RNA polymerase. Taking into account the troubles we had with the construction of the plasmids with the T7-ccdB insert, we can almost certainly conclude that despite this the cells did experience hindering of the CcdB toxin. Often we obtained no colonies after transformation and if we did, there were mutations in the toxic part of the ccdB gene (see further). We presume that this can only be caused if the ccdB was transcribed by other polymerases who are not specific for the T7 promoter. This probably only occurs at very low frequencies, but as in these strains there is no CcdA present at all even the lowest amount of working CcdB can be lethal for these cells. Clone Manager and I-TASSERIn order to examine the effect of these mutations on the protein structure, we determined the amino acid sequence of the mutated plasmid 8 + pSB6A1-T7ccdB, 2 mM IPTG day 2, Step using Clone Manager:
MAQFKVYTYKRESRYRLFVDVQSDIIDTPGRRMVIPLASARLLSDKVSRELYPVVHIGDESWRMMTTDMASVPVSVI
At the position of the Tryptophan residue responsible for interaction with Arg462 and consequently essential for toxic activity, there is now a Glycine residue. Also the following two amino acids essential for toxicity are mutated. Furthermore chances are that the following part of the protein will sterically hinder interaction with DNA gyrase. As a conclusion this amino acid sequence is a good indication that this mutated CcdB protein won’t be able to execute its toxic function.
To further investigate the consequences of the frameshift we used I-TASSER to predict the structure of this protein, resulting in following top 5 of predicted models:
Note: C-score is typically in the range of [-5,2], where a C-score of higher value signifies a model with a high confidence and vice-versa.  Further research perspectives
Assemble T7-ccdB plasmid inside CcdB-survival strains SourcesDao-Thi, M.H. et al. Molecular basis of gyrase poisoning by the addiction toxin CcdB. Journal of Molecular Biology 348, 1091-1102 (2005).Couturier, M., Bahassi, E. & Van Melderen, L. Bacterial death by DNA gyrase poisoning. Trends in Microbiology 6, 269-275 (1998). Corzett, C.H., Goodman, M.F. & Finkel, S.E. Competitive Fitness During Feast and Famine: How SOS DNA Polymerases Influence Physiology and Evolution in Escherichia coli. Genetics 194, 409-420 (2013).
|

Tweets van @iGEM_UGent |
|||||||||||||||||||||||||||||||||
















|
|




