 "
"
Team:Tokyo Tech/Project/M13 Shuriken
From 2013.igem.org
| Line 43: | Line 43: | ||
<h1>4. Model system for inducible phage release</h1><h2> | <h1>4. Model system for inducible phage release</h1><h2> | ||
We constructed a model system for inducible phage release by regulation of <i>g2p</i> expression. Genome DNA of this engineered phage, shown in Fig. 2-2-6, needs two functions. One is inducible expression of <i>g2p</i> protein. We thus designed to replace the promoter for <i>g2p</i> protein with <i>lux</i> promoter. Note that we used 3OC6HSL, not 3OC12HSL , in this model experiment. The other is maintenance of the genome DNA in the absence of <i>g2p</i> expression. We combined M13 genome double stranded DNA with pSB3K3 backbone. | We constructed a model system for inducible phage release by regulation of <i>g2p</i> expression. Genome DNA of this engineered phage, shown in Fig. 2-2-6, needs two functions. One is inducible expression of <i>g2p</i> protein. We thus designed to replace the promoter for <i>g2p</i> protein with <i>lux</i> promoter. Note that we used 3OC6HSL, not 3OC12HSL , in this model experiment. The other is maintenance of the genome DNA in the absence of <i>g2p</i> expression. We combined M13 genome double stranded DNA with pSB3K3 backbone. | ||
| - | [[Image:Titech2013_M13_newpart.png| | + | [[Image:Titech2013_M13_newpart.png|600px|thumb|center|Fig. 2-2-6. Design of phage DNA for inducible phage release, pSB3K3 backbone is required for maintenance of the DNA without inducer.]] |
<p> | <p> | ||
Firstly, we confirmed that M13 genome with two modifications related to our design still had plaque forming activity. One is replacement of the promoter for <i>g2p</i> protein with a constitutive promoter, PlacI<sup>q</sup> ([http://parts.igem.org/Part:BBa_I14032 BBa_I14032]). The other is accommodation of pSB3K3 backbone. Even though the plasmid has two different types of replication origins, M13 origin and pSB3 origin, this plasmid ([http://parts.igem.org/Part:BBa_K1139020 BBa_K1139020]) formed plaque(Fig. 2-2-7). In contrast, construction intermediates without a promoter upstream of <i>g2p</i> coding sequence (Promoterless-M13 + Plac: [http://parts.igem.org/Part:BBa_K1139018 BBa_K1139018], Promoterless-M13 + Plac-GFP: [http://parts.igem.org/Part:BBa_K1139022 BBa_K1139022]) could not form plaque ([https://2013.igem.org/Team:Tokyo_Tech/Experiment/pSB-M13_Plasmid_Assay#1._Introduction See more about experiment: pSB-M13 Plasmid Assay]). | Firstly, we confirmed that M13 genome with two modifications related to our design still had plaque forming activity. One is replacement of the promoter for <i>g2p</i> protein with a constitutive promoter, PlacI<sup>q</sup> ([http://parts.igem.org/Part:BBa_I14032 BBa_I14032]). The other is accommodation of pSB3K3 backbone. Even though the plasmid has two different types of replication origins, M13 origin and pSB3 origin, this plasmid ([http://parts.igem.org/Part:BBa_K1139020 BBa_K1139020]) formed plaque(Fig. 2-2-7). In contrast, construction intermediates without a promoter upstream of <i>g2p</i> coding sequence (Promoterless-M13 + Plac: [http://parts.igem.org/Part:BBa_K1139018 BBa_K1139018], Promoterless-M13 + Plac-GFP: [http://parts.igem.org/Part:BBa_K1139022 BBa_K1139022]) could not form plaque ([https://2013.igem.org/Team:Tokyo_Tech/Experiment/pSB-M13_Plasmid_Assay#1._Introduction See more about experiment: pSB-M13 Plasmid Assay]). | ||
Revision as of 03:18, 28 September 2013

M13 "Shuriken"
Contents |
1. Introduction
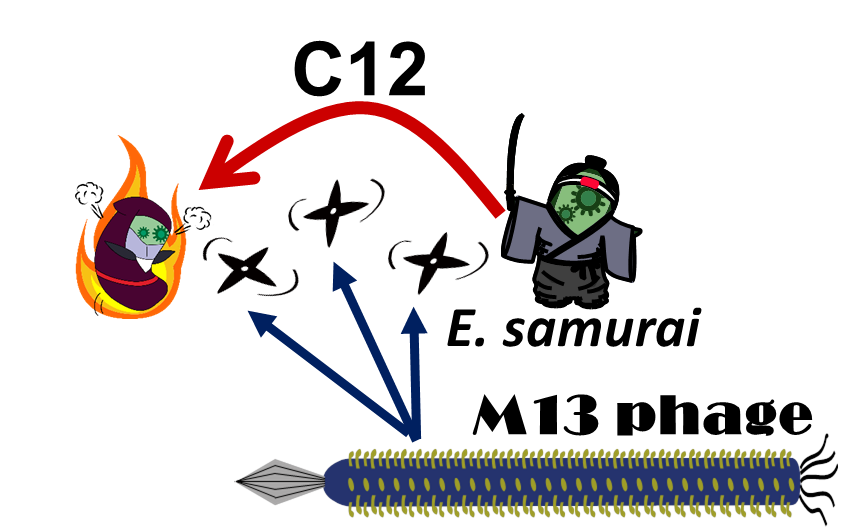
 Fig. 2-2-1. M13 phage play the role of “shuriken”.
Fig. 2-2-1. M13 phage play the role of “shuriken”.

E. ninja throws "shuriken" to attack E. samurai in response to 3OC12HSL, an intercellular molecule. In our story, M13 phages are the shuriken. We confirmed that the system of AHL-dependent release and infection of M13 phage shuriken properly worked, using plaque forming assay.

We constructed the plasmid for the system of inducible release of M13 phage (Fig. 2-2-2). In this system, E. coli releases the phage in response to induction. We inserted the sequence amplified from M13mp18 vector to pSB3K3 backbone. Next, we replaced the promoter upstream of g2p, which is concerned with the release of M13 phage.
Our assay revealed that the plasmid we designed still had plaque forming activity (pSB-M13 Plasmid Assay).
 Fig. 2-2-3. The analysis of the distribution of the plaques by using ImageJ |
 Fig. 2-2-4. The distribution histogram of the plaques |
We confirmed whether the release of the M13 phage particle depends on the induction of g2p expression. Fig. 2-2-3 and Fig. 2-2-4 are the results of the assay. We analyzed the distribution of plaques by using ImageJ (Fig. 2-2-3). This histogram shows distance from the piece of paper in a horizontal axis, and shows number of plaques over distance squared, which means the rate of plaque formation, in a vertical axis (Fig. 2-2-4). This histogram shows that the inducible release of M13 phage is realized (inducible Plaque Forming Assay).
2. Phage life cycle dependent on g2p expression
In natural M13 phage life cycle, g2p expression is required for production of a single stranded DNA (ssDNA) and thus required for phage release. Nicking on a double stranded DNA by G2p nickase triggers a series of reactions to amplify the phage ssDNA. In the presence of the ssDNA, the coat proteins are assembled around it (See more about phage assembly). [1][2]
3. Inducible M13 phage release
in the whole circuit design
In our story, E. ninja releases the phage particles, only when E. ninja is in the Attack state induced by 3OC12HSL. In the whole circuit (Fig. 2-2-5), g2p expression is indirectly regulated by 3OC12HSL via cI expression.

4. Model system for inducible phage release
We constructed a model system for inducible phage release by regulation of g2p expression. Genome DNA of this engineered phage, shown in Fig. 2-2-6, needs two functions. One is inducible expression of g2p protein. We thus designed to replace the promoter for g2p protein with lux promoter. Note that we used 3OC6HSL, not 3OC12HSL , in this model experiment. The other is maintenance of the genome DNA in the absence of g2p expression. We combined M13 genome double stranded DNA with pSB3K3 backbone.

Firstly, we confirmed that M13 genome with two modifications related to our design still had plaque forming activity. One is replacement of the promoter for g2p protein with a constitutive promoter, PlacIq ([http://parts.igem.org/Part:BBa_I14032 BBa_I14032]). The other is accommodation of pSB3K3 backbone. Even though the plasmid has two different types of replication origins, M13 origin and pSB3 origin, this plasmid ([http://parts.igem.org/Part:BBa_K1139020 BBa_K1139020]) formed plaque(Fig. 2-2-7). In contrast, construction intermediates without a promoter upstream of g2p coding sequence (Promoterless-M13 + Plac: [http://parts.igem.org/Part:BBa_K1139018 BBa_K1139018], Promoterless-M13 + Plac-GFP: [http://parts.igem.org/Part:BBa_K1139022 BBa_K1139022]) could not form plaque (See more about experiment: pSB-M13 Plasmid Assay).

We then confirmed that replacement of the g2p promoter with lux promoter, using as a model inducible promoter, accomplished inducible phage release. For a plaque forming assay, we used a plasmid with lux promoter upstream of g2p coding sequence (Plux-M13-Plac-GFP: [http://parts.igem.org/Part:BBa_K1139021 BBa_K1139021]) (See more about DNA construction). First, we cultured E. coli which is introduced our new part ([http://parts.igem.org/Part:BBa_K1139021 BBa_K1139021]), and added AHL, which induces the phage release. We then centrifuged the culture and obtained phage particles. Finally, we added the phage particles and lawn culture (E. coli containing pSB6A1-Ptet-luxR) to the soft agar, and poured it on an agar plate. Also, to make a concentration gradient of the inducer (AHL), we put a piece of filter paper on which dropped the AHL solution.

The result shows that the plaques are formed only when the inducer exists in the medium at appropriate concentration. On the lawn, we firstly put, two pieces of filter paper with or without AHL (Fig. 2-2-9 A). Plaque formation was dependent on distance from the paper with AHL. For further analysis, we thus put only a piece of filter paper with AHL in the center of the plate (Fig. 2-2-9 B). The distribution of the plaques has an optimum value, depending on the distance from the piece of filter paper with AHL (Fig. 2-2-10).
At circular region with appropriate distance from the piece of filter paper, the expression of g2p is so activated that the phage particles cannot be produced efficiently. The reason why no plaques were formed near the piece of the filter paper is the production of the coat proteins largely exceeds that of the single stranded DNA. In contrast, far from the piece of filter paper, the expression of g2p is so poor that the phage particles cannot be produced efficiently, because the production of the single stranded DNA isn’t enough to produce phage particles.
These result shows that the inducible release of M13 phage is realized; our plan was achieved (See more about experiment : inducible Plaque Forming Assay).

5. Application
We achieved the DNA messaging system using M13 phage which is released by promoter induction. You can apply our DNA messaging system to following two ideas because you utilize various stimuli for induction.
Drew Endy's group recently reported that they achieved the cell-cell communication system in which DNA is regarded as message [3] (Fig. 2-2-11). In this system, phages, which transmit DNA to receiver cell, were released by a limited stimulation: addition induction of a helper phage. On the other hand, if you apply our DNA messaging system, you can make DNA messaging system more versatile and complex because you utilize various stimuli, not limiting the inducer of phage release to a helper phage [4] (Fig. 2-2-12).


It is also important to research infectious spectrum, what kinds of bacteria phage can infect, before we use phage for bioremediation. In future, we could use a phage as carrier of DNA transfection for bioremediation. M13 phage can be a model system for such case. If you use M13 phage for transfecting to bacteria living in the environment, we need to research what kinds of bacteria can be the host of M13 phage using a sample of the soil. The gene coding fluorescence protein in the engineered phage we designed is useful when you research the host of M13 phage. When M13 phage including the genetic circuit cording fluorescence protein infects bacteria, they shine fluorescence. Because the fluorescence intensity depends on the kind of infected cell, you can separate these kinds of bacteria by cell sorter. Also, you can analyze the genome of infected bacteria by single cell genomics. As a similar study, Nojiri’s group and Shintani’s group reported that they can transfect plasmid DNA to uncultivable bacteria in soil. [5][6] They use conjugation for transfection of DNA in this research. You can make the idea more versatile by applying our idea because you can also transfect DNA to bacteria which can be the host of M13 phage (Fig. 2-2-13). Moreover, you could identify the strain which would be the host of M13 phage only on a specific condition because you can control the phage release with knowledge of synthetic biology. For example, if you could find the strain which fluoresces only when AHL exists, the strain would be the host on the condition that AHL exists (Fig. 2-2-14, 2-2-15).


