 "
"
Team:UGent/Results
From 2013.igem.org
| Line 147: | Line 147: | ||
<ul> | <ul> | ||
<li>We subtracted the OD of all the wells with the average OD of the blank and did the same for the fluorescence intensity. This was done to standardize the measurements. </li> | <li>We subtracted the OD of all the wells with the average OD of the blank and did the same for the fluorescence intensity. This was done to standardize the measurements. </li> | ||
| - | <li>Because we used many different plates (and thus some strains were spread over two or more plates), we added a reference strain (strain 8) to all the plates. This strain contains one copy of our construct and thus should give the same F/OD in every well. We used various statistical tests to check if there was a significant difference in average F/OD between the plates of one strain. If these tests resulted in a significant difference of the 8 strain, we could not compare the measurements. Luckily this only happened for a few strains. The extensive execution of these tests can be checked in <a href="https://static.igem.org/mediawiki/2013/d/db/UGent_2013_statistics.pdf" target="_blank">this pdf file</a>.</li> | + | <li>Because we used many different plates (and thus some strains were spread over two or more plates), we added a reference strain (strain 8) to all the plates. This strain contains one copy of our construct and thus should give the same F/OD in every well. We used various statistical tests to check if there was a significant difference in average F/OD between the plates of one strain. If these tests resulted in a significant difference of the 8 strain, we could not compare the measurements. Luckily this only happened for a few strains. The extensive execution of these tests can be checked in <a href="https://static.igem.org/mediawiki/2013/d/db/UGent_2013_statistics.pdf" target="_blank">this pdf file</a>.</li></ul> |
<br><br> | <br><br> | ||
Thereafter we calculated the F/OD for every well. If the data showed some abnormalities, we tested them for outliers. If the values of the outliers were significantly different than the values of the rest of the data, we excluded these observations and did not use them in our further analysis. | Thereafter we calculated the F/OD for every well. If the data showed some abnormalities, we tested them for outliers. If the values of the outliers were significantly different than the values of the rest of the data, we excluded these observations and did not use them in our further analysis. | ||
Revision as of 00:16, 5 October 2013
 |
|||||||||||||||||||||||
|
Strains with ccdA-gfp constructThe following strain containing the construct to be duplicated (ccdA-gfp) was constructed (KI experiment 1):
We have two versions of this strain: 5 and 8. The two strains were used in our further experiments as to test possible differences between the two versions. Plasmids containing T7-ccdB
We constructed plasmid pSB6A1-T7ccdB for use in CIChE. Through this plasmid, pressure can be put on the CIChE strains simply by adding IPTG.
CIChE Strains
The plasmids and strains mentioned above were used in transformations (experiment 3) to obtain strains containing all elements necessary to perform chromosomal evolution. The following strains were constructed:
UV Test Phage transduction was used to perform the deletion of the recA gene after chromosomal evolution. To check whether the gene was successfully knocked out, an UV test was developed. The UV light puts a certain amount of stress on the bacterial cells. In respons to this stress, an SOS pathway is triggered in which recA plays an important role. Cells in which the recA gene is deleted will not survive on the UV-treated plate. Colonies in which recA was successfully deleted still can be found on a non-treated backup plate.
CIChE
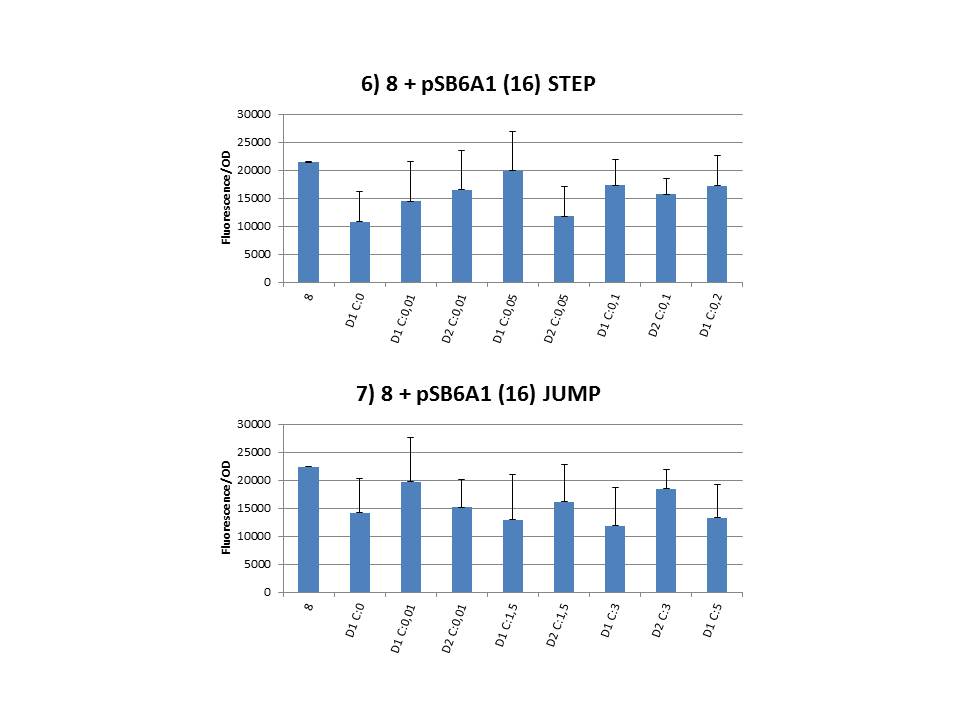
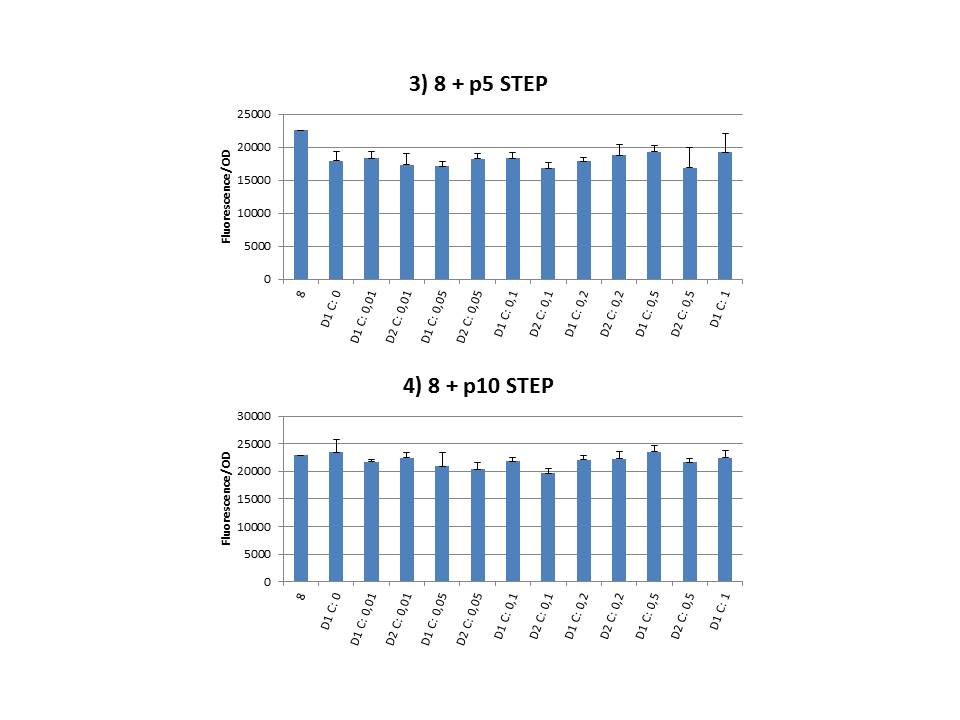
The practical execution of CIChE was as follows: a strain was inoculated with the right concentration of IPTG (day 1 = D1), these strains were grown overnight at 37°C. The next day the strains were inoculated with the same IPTG concentration (day 2 = D2) and with an increased IPTG level (D1). IPTG concentration was raised with a small “step” or with a larger “jump”. These strains were also grown overnight at 37°C. This was repeated over the next days. Because the outcome was difficult to predict, a lot of different IPTG concentrations and ranges were tested.
Thereafter we calculated the F/OD for every well. If the data showed some abnormalities, we tested them for outliers. If the values of the outliers were significantly different than the values of the rest of the data, we excluded these observations and did not use them in our further analysis. Finally we plotted the F/OD in function of the IPTG concentration. The results can be seen in figure 1-9 (you can click on the graphs to enlarge). In every graph the average F/OD of strain 8 is added as reference. The average is calculated using the results of 8 corresponding to the correct plates. If our model for CIChE works, our strains (containing more gfp copies) should have an average F/OD higher than the one of strain 8. We also expected that the F/OD would rise with increasing IPTG concentrations. Looking at the graphs we see that most of our strains result in an average F/OD lower than or about the same as the average result of strain 8. This is not at all what we expected/hoped. According to the obtained data, we must conclude that no duplication of the construct occurred, hence no chromosomal evolution was achieved. However, we have a pretty good idea what lead to these results. Our hypothesis can be read below.
Why are we having so much trouble?For the construction of our plasmid containing the ccdB gene we chose to work with a T7 promoter for the ccdB gene as to avoid leaky expression. Taking into account the troubles we had with the construction of the plasmids with the T7-ccdB insert we can almost certainly conclude that this promoter does still show significant leaky expression, as the toxin gene obviously hindered the cells although the promoter was not induced. Often we obtained no colonies after transformation and if we did, there were mutations in the ccdB gene. We performed CIChE with the plasmid we constructed ourselves (pSB6A1-T7ccdB) as well as with the original plasmids from which we constructed this part: p5SpFRT-T7ccdB, p10SpFRT-T7ccdB and p20SpFRT-T7ccdB. According to the GFP measurements we did, none of these resulted in duplication of the ccdA-gfp construct in the genome. We presume that this is caused by the malfunction of CcdB caused by mutations. But how is it possible that there are so many of these mutations? It is known from previous research that the part essential for CcdB’s toxic activity is situated at the last three amino acids (Trp-Gly-Ile). Interaction between CcdB and the DNA gyrase A subunit (GyrA) is situated at Trp99 from CcdB and Arg462 of GyrA59 (the 59 kDa amino-terminal breaking-rejoining domain of GyrA). Random mutations in the final part of the ccdB gene affecting this toxic site will consequently provide these cells with a great growth advantage over cells with intact ccdB genes. We assume this is a probable reason why mutation frequency of the ccdB gene seems to be so high in our project. Furthermore, in the absence of its antidote CcdA, CcdB causes reduced DNA synthesis, activation of the SOS pathway, cell filamentation and eventually cell death. One of the aspects of this SOS pathway is the enhancment of capacity of mutagenesis, such as point and frameshift mutations. So if for example there is leaky expression of the promoter and CcdB is being formed without a sufficient amount of CcdA present, the SOS pathway will be activated causing higher mutation frequencies. We postulate that these mutations can also affect the ccdB gene itself and thus restoring ‘normal’ cell life. In combination with what we discussed in the previous paragraph this could explain the amount of mutations we had to deal with over the course of our project.
Randomly we chose a strain at a point during the CIChE experiment (8 + pSB6A1-T7ccdB, 2 mM IPTG day 2, Step) and let the plasmid get sequenced. When we compared the ccdB sequence to the native one we saw our plasmid had a deletion of a cytosine residue at position 297 (out of a total of 306 base pairs). This causes a frameshift, resulting in a prolonged protein of 154 amino acids instead of the native structure with 102 amino acids. (Note: CcdB normally contains 101 amino acids, but the sequence we used has an extra Alanine after the start codon for cloning purposes.)
To be continued...
 Further research perspectives
Assemble T7ccdB plasmid inside CcdB-survival strains SourcesDao-Thi, M.H. et al. Molecular basis of gyrase poisoning by the addiction toxin CcdB. Journal of Molecular Biology 348, 1091-1102 (2005).Couturier, M., Bahassi, E. & Van Melderen, L. Bacterial death by DNA gyrase poisoning. Trends in Microbiology 6, 269-275 (1998). Corzett, C.H., Goodman, M.F. & Finkel, S.E. Competitive Fitness During Feast and Famine: How SOS DNA Polymerases Influence Physiology and Evolution in Escherichia coli. Genetics 194, 409-420 (2013).
|

Tweets van @iGEM_UGent |
||||||||||||||||||||||











|
|




