 "
"
Team:SydneyUni Australia/Project/Results
From 2013.igem.org
(Difference between revisions)
Jbergfield (Talk | contribs) |
C.Squirrel (Talk | contribs) |
||
| Line 29: | Line 29: | ||
text-decoration: none; | text-decoration: none; | ||
text-color: #82CA9C; | text-color: #82CA9C; | ||
| + | } | ||
| + | |||
| + | .pictext { | ||
| + | height: 100px; | ||
| + | margin: 10px 0px; | ||
| + | width: 100% | ||
} | } | ||
.pictextl { | .pictextl { | ||
float: left; | float: left; | ||
| - | width: | + | width: 70%; |
vertical-align: middle; | vertical-align: middle; | ||
| - | margin-left: | + | margin-left: 0%; |
} | } | ||
| Line 62: | Line 68: | ||
</ul> | </ul> | ||
<li>We showed that ToMO can begin degradation of DCA through an assay for chloride ions (link to protocol) released as DCA is converted to chloroacetaldehyde. To our knowledge this has’t been shown by anyone else before. </li> | <li>We showed that ToMO can begin degradation of DCA through an assay for chloride ions (link to protocol) released as DCA is converted to chloroacetaldehyde. To our knowledge this has’t been shown by anyone else before. </li> | ||
| - | + | ||
| - | <div class="pictextl" style="height: 100px; line-height: 100px">Table of results from Cl- assay showing how awesome ToMO is:</div> | + | <div class="pictext"> |
| - | + | <div class="pictextl" style="height: 100px; line-height: 100px">Table of results from Cl- assay showing how awesome ToMO is:</div> | |
| - | + | <div class="pictextr" style="height: 100px;"> | |
| - | + | <center> | |
| - | + | <a href="https://static.igem.org/mediawiki/2013/a/a3/Sydney_Australia_2013_Results_ToMOonDCA.jpg" rel="ibox" title="ToMO Cl- Assay"> | |
| - | + | <img src="https://static.igem.org/mediawiki/2013/a/a3/Sydney_Australia_2013_Results_ToMOonDCA.jpg" height="100"> | |
| - | + | </a> | |
| - | + | </center> | |
| - | + | </div> | |
| + | </div> | ||
| + | |||
| + | |||
<li>This is pretty cool, but during the middle of the year we decided to try synthesising the whole pathway rather than building it by conventional cloning. The length of the ToMO gene cluster meant it was too expensive for us to continue working with it.</li> | <li>This is pretty cool, but during the middle of the year we decided to try synthesising the whole pathway rather than building it by conventional cloning. The length of the ToMO gene cluster meant it was too expensive for us to continue working with it.</li> | ||
| - | + | ||
| - | <div class="pictextl" style="height: 100px; line-height: 100px" | + | <div class="pictext"> |
| - | + | <div class="pictextl" style="height: 100px; line-height: 100px">E. coli expressing ToMO converts indol to an indo-coloured compound:</div> | |
| - | + | <div class="pictextr" style="height: 100px;"> | |
| - | + | <center> | |
| - | + | <a href="https://static.igem.org/mediawiki/2013/9/97/SydneyUni2013_Results_TomoIndigo.jpg" rel="ibox" title="ToMO Cl- Assay"> | |
| - | + | <img src="https://static.igem.org/mediawiki/2013/9/97/SydneyUni2013_Results_TomoIndigo.jpg" height="100"> | |
| - | + | </a> | |
| + | </center> | ||
| + | </div> | ||
</div> | </div> | ||
| + | |||
</ul> | </ul> | ||
<div class="unlink">Gibson Assembly Was Problematic</div> | <div class="unlink">Gibson Assembly Was Problematic</div> | ||
| Line 89: | Line 101: | ||
<li> Transformation </li> | <li> Transformation </li> | ||
<ul> | <ul> | ||
| - | <li>We spent a week <b>(which week? link to calendar)</b> optimising the transformation of our Gibson Assembly reaction product ( | + | <li>We spent a week <b>(which week? link to calendar)</b> optimising the transformation of our Gibson Assembly reaction product <b>(link to explanation of Gibson Assembly and our pathway)</b>. We initially tried transformation into E. Coli EPI300 and yielded no transformants. We suspected that there may be a metabolic burden or harm to cells carrying our correctly assembled product, due to the strong constitutive expression of our designed promoter, Psyn <b>(link to design of gBlocks, Psyn explanation)</b>. To account for this we tried transforming into E. Coli EPI400, which carries plasmids at low copy-number with an inducible increase in copy-number. We also tried incubating and growing cells at room temperature to lessen their growth rate, so that they might be able to better cope with any possible toxicity of the transformed Gibson Assembly reaction product. Neither of these were successful, however, we were able to screen 87 clones by transforming into a different strain, E. Coli TOP10.</li> |
</ul> | </ul> | ||
<li> Screening </li> | <li> Screening </li> | ||
| Line 118: | Line 130: | ||
<li> Modularity </li> | <li> Modularity </li> | ||
<ul> | <ul> | ||
| - | <li> Consider the parable of the watchmakers: <br><br> There once were two watchmakers, named Hora and Tempus, who manufactured very | + | <li> Consider the parable of the watchmakers: <br><br> There once were two watchmakers, named Hora and Tempus, who manufactured very fine watches. Both of them were highly regarded, and the phones in their workshops rang frequently - new customers were constantly calling them. However, Hora prospered, while Tempus became poorer and poorer and ?nally lost his shop.What was the reason?<br><br>The watches the men made consisted of about 1,000 parts each. Tempus had so constructed his that if he had one partly assembled and had to put it down - to answer the phone say - it immediately fell to pieces and had to be reassembled from the elements. The better the customers liked his watches, the more they phoned him, the more difficult it became for him to find enough uninterrupted time to finish a watch.<br><br>The watches that Hora made were no less complex than those of Tempus. But he had designed them so that he could put together subassemblies of about ten elements each. Ten of these subassemblies, again, could be put together into a larger subassembly; and a system of ten of the latter sub-assemblies constituted the whole watch. Hence, when Hora had to put down a partly assembled watch in order to answer the phone, he lost only a small part of his work, and he assembled his watches in only a fraction of the man-hours it took Tempus.<br><br>H.A. Simon, The Architecture of Complexity, 1962.</li> |
<li>The sequences we had synthesised as gBlocks by IDT were designed so that they could only be assembled in the whole DCA-degradation pathway, rather than as parts within pSB1C3 which could then be assembled piece-by-piece. This meant that our success relied on the correct assembly of the entire pathway, and when this failed, that we were unable to access parts of the pathway (without PCR assembly, or ordering new, complementary gBlocks).</li> | <li>The sequences we had synthesised as gBlocks by IDT were designed so that they could only be assembled in the whole DCA-degradation pathway, rather than as parts within pSB1C3 which could then be assembled piece-by-piece. This meant that our success relied on the correct assembly of the entire pathway, and when this failed, that we were unable to access parts of the pathway (without PCR assembly, or ordering new, complementary gBlocks).</li> | ||
</ul> | </ul> | ||
| Line 142: | Line 154: | ||
<ul> | <ul> | ||
<li>We designed primers specifically for amplifying dhlB and dhlA, while removing a forbidden EcoRI site between them. Design with the primers allowed us to try cloning each gene by itself and also together into the shipping vector pSB1C3. | <li>We designed primers specifically for amplifying dhlB and dhlA, while removing a forbidden EcoRI site between them. Design with the primers allowed us to try cloning each gene by itself and also together into the shipping vector pSB1C3. | ||
| - | < | + | |
| - | + | <div class="pictext"> | |
| + | <div class="pictextl"><b>DESCRIPTION</b></div> | ||
| + | <div class="pictextr"> | ||
| + | <center> | ||
| + | <a href="https://static.igem.org/mediawiki/2013/1/1c/SydneyUni2013_Results_Assembly_Amplification.jpg" rel="ibox" title="ToMO Cl- Assay"> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/1/1c/SydneyUni2013_Results_Assembly_Amplification.jpg" height="100"> | ||
| + | </a> | ||
| + | </center> | ||
| + | </div> | ||
| + | </div> | ||
| + | |||
</ul> | </ul> | ||
<li> Cloning </li> | <li> Cloning </li> | ||
<ul> | <ul> | ||
| - | < | + | |
| - | + | <div class="pictext"> | |
| + | |||
| + | <div class="pictextl">We cloned the PCR fragments into pSB1C3 and transformed the ligation product. We were greatly assisted by ligating into a <a href="http://parts.igem.org/Part:BBa_J04450">BBa_J04450</a>, extracted from the <a href="http://parts.igem.org/Help:Protocols/Linearized_Plasmid_Backbones">linearised plasmid pSB1C3</a> in the Distribution Kit, which provided a neat red-white screen.</div> | ||
| + | <div class="pictextr"> | ||
| + | <center> | ||
| + | <a href="https://static.igem.org/mediawiki/2013/5/55/SydneyUni2013_Results_Assembly_Cloning_Screen.jpg" rel="ibox" title="ToMO Cl- Assay"> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/5/55/SydneyUni2013_Results_Assembly_Cloning_Screen.jpg" height="100"> | ||
| + | </a> | ||
| + | </center> | ||
| + | </div> | ||
| + | </div> | ||
| + | |||
| + | |||
| + | |||
<li>After PCR screening of the junctions between our parts and pSB1C3, we extracted the plasmids from a few that looked OK for further confirmation and submission to the iGEM HQ. | <li>After PCR screening of the junctions between our parts and pSB1C3, we extracted the plasmids from a few that looked OK for further confirmation and submission to the iGEM HQ. | ||
| - | + | ||
| + | <div class="pictext"> | ||
| + | <div class="pictextl"><b>DESCRIPTION</b></div> | ||
| + | <div class="pictextr"> | ||
| + | <center> | ||
| + | <a href="https://static.igem.org/mediawiki/2013/5/54/SydneyUni2013_Results_Assembly_Cloning_Digest.jpg" rel="ibox" title="ToMO Cl- Assay"> | ||
| + | <img src="https://static.igem.org/mediawiki/2013/5/54/SydneyUni2013_Results_Assembly_Cloning_Digest.jpg" height="100"> | ||
| + | </a> | ||
| + | </center> | ||
| + | </div> | ||
| + | </div> | ||
| + | |||
<li><b> DOUBLE DIGEST??? PCR??? Someone decide and write something I don't have time. </b></li> | <li><b> DOUBLE DIGEST??? PCR??? Someone decide and write something I don't have time. </b></li> | ||
</ul> | </ul> | ||
Revision as of 01:57, 28 September 2013


Project Results
ToMO degrades DCA
- Early in our project we needed to find a suitable monooxygenase to begin degradation of DCA by one of the two degradation pathways.
MISSING PICTURES - Toluene-o-xylene monooxygenase (ToMO) from Pseuodomonas stutzeri OX1 has been shown to oxidise xylenes, toluene, benzene, styrene, napthalene (Bertoni et al, 1996) as well as tetrachloroethene, trichloroethene, dichloroethene and vinyl chloride (Shim et al, 2001). The enzyme was optimised for chlorinated ethene degradation (Varder & Wood, 2005), and gifted to our host lab in the plasmid pBS(Kan)ToMO.
- Bertoni, G., Bolognese, F., Galli, E., & Barbieri, P. (1996). Cloning of the genes for and characterization of the early stages of toluene and o-xylene catabolism in Pseudomonas stutzeri OX1. Applied and environmental microbiology, 62(10), 3704-3711.
- Shim, H., Ryoo, D., Barbieri, P., & Wood, T. (2001). Aerobic degradation of mixtures of tetrachloroethylene, trichloroethylene, dichloroethylenes, and vinyl chloride by toluene-o-xylene monooxygenase of Pseudomonas stutzeri OX1. Applied microbiology and biotechnology, 56(1-2), 265-269.
- Vardar, G., & Wood, T. K. (2005). Protein engineering of toluene-o-xylene monooxygenase from Pseudomonas stutzeri OX1 for enhanced chlorinated ethene degradation and o-xylene oxidation. Applied microbiology and biotechnology, 68(4), 510-517.
- We showed that ToMO can begin degradation of DCA through an assay for chloride ions (link to protocol) released as DCA is converted to chloroacetaldehyde. To our knowledge this has’t been shown by anyone else before.
- This is pretty cool, but during the middle of the year we decided to try synthesising the whole pathway rather than building it by conventional cloning. The length of the ToMO gene cluster meant it was too expensive for us to continue working with it.
Table of results from Cl- assay showing how awesome ToMO is:

E. coli expressing ToMO converts indol to an indo-coloured compound:

Gibson Assembly Was Problematic
- Progress
- Transformation
- We spent a week (which week? link to calendar) optimising the transformation of our Gibson Assembly reaction product (link to explanation of Gibson Assembly and our pathway). We initially tried transformation into E. Coli EPI300 and yielded no transformants. We suspected that there may be a metabolic burden or harm to cells carrying our correctly assembled product, due to the strong constitutive expression of our designed promoter, Psyn (link to design of gBlocks, Psyn explanation). To account for this we tried transforming into E. Coli EPI400, which carries plasmids at low copy-number with an inducible increase in copy-number. We also tried incubating and growing cells at room temperature to lessen their growth rate, so that they might be able to better cope with any possible toxicity of the transformed Gibson Assembly reaction product. Neither of these were successful, however, we were able to screen 87 clones by transforming into a different strain, E. Coli TOP10.
- Screening
- We screened 87 clones for the presence of dhlB (link to calendar), a gene responsible for the breakdown of chloroacetate in our pathway, by incubating resting cells with chloroacetate and chloride assay (link to protocol). A few clones from each type of pathway looked promising, so we proceeded to extract plasmids from these for further investigation.
- GET PICTURES FOR ABOVE
- Later on (link to calendar, it was about a week later, after a set of primers had arrived), we also tried screening for dhlB by PCR. We amplicon of interest would have spanned only a single overlap during Gibson Assembly, yet we failed to find a single clone containing the assembled (or misassembled) gene dhlB.
- GET PICTURES FOR ABOVE
- Plasmid Preps
- None of the clones from which we extracted plasmids contained the correctly assembled insert. By PCR and diagnostic restriction digests on these plasmids we were able to distinguish two different misassembled versions of our desired Gibson Assembly reaction product.
- GET PICTURES FOR ABOVE
- PCR
- We thought it could have been possible to assemble our entire pathway from smaller fragments salvaged from our Gibson Assembly reaction product. This proved impossible, presumably due to the extent of heterogenous template including both correctly and incorrectly assembled gBlocks in the Gibson Assembly reaction product. It may have been possible to do something similar using IDT’s gBlocks as template, however, this was not possible as we’d used up all of one of gBlocks during a second Gibson Assembly.
- GET PICTURES FOR ABOVE
- Lessons
- Constitutive Expression
- We suspect that some of the genes we tried to assemble (eg, p450, Nishino et al, 2013, dropboxed in ‘reading’) can harm the cells they're expressed in. If this is the case, then by a sort of natural screening we were only able to find colonies on plates that contain misassembled gBlocks that did not express these genes.
- Upon reflection, we approached the assembly of our pathway with an almost child-like ignorance and optimism. Our promoter was specifically designed to maximise expression of our construct, as if ‘the more pollutant degrading genes, the better’. If the hypothesis above is correct, then we might have had more success with an inducible promoter.
- Modularity
- Consider the parable of the watchmakers:
There once were two watchmakers, named Hora and Tempus, who manufactured very fine watches. Both of them were highly regarded, and the phones in their workshops rang frequently - new customers were constantly calling them. However, Hora prospered, while Tempus became poorer and poorer and ?nally lost his shop.What was the reason?
The watches the men made consisted of about 1,000 parts each. Tempus had so constructed his that if he had one partly assembled and had to put it down - to answer the phone say - it immediately fell to pieces and had to be reassembled from the elements. The better the customers liked his watches, the more they phoned him, the more difficult it became for him to find enough uninterrupted time to finish a watch.
The watches that Hora made were no less complex than those of Tempus. But he had designed them so that he could put together subassemblies of about ten elements each. Ten of these subassemblies, again, could be put together into a larger subassembly; and a system of ten of the latter sub-assemblies constituted the whole watch. Hence, when Hora had to put down a partly assembled watch in order to answer the phone, he lost only a small part of his work, and he assembled his watches in only a fraction of the man-hours it took Tempus.
H.A. Simon, The Architecture of Complexity, 1962. - The sequences we had synthesised as gBlocks by IDT were designed so that they could only be assembled in the whole DCA-degradation pathway, rather than as parts within pSB1C3 which could then be assembled piece-by-piece. This meant that our success relied on the correct assembly of the entire pathway, and when this failed, that we were unable to access parts of the pathway (without PCR assembly, or ordering new, complementary gBlocks).
- Plans
- Replacement of gBlocks
- By replacing a single gBlock (link to gBlock design, I’m talking about iGEMBLOCK1, which contains an overlap with pSB1C3, Psyn, an RBS and the beginning of the gene aldA) it might be possible to find correctly assembled Gibson products by substituting our strong constitutive promoter Psyn with a repressible promoter.
- With four new gBlocks, it might be possible to assemble some of the important genes in our pathway (aldA, p450, adh1b1) in a BioBrick vector, so that they could then be subsequently assembled piecewise.
- PCR Assembly
- It might be possible to PCR amplify genes directly from our gBlocks as template rather than our Gibson Assembly reaction product. Alternatively, with the design of new primers, the genes of interest might be amplified from gBlocks for cloning into a BioBrick vector. With either of these options, sequence fidelity may be an issue.
Assembly of dhlB-dhlA in pSB1C3
- While struggling with Gibson Assembly of our whole pathway we turned to the extraction, cloning and characterisation of two parts in our pathway, dhlB and dhlA (link to pathway or pic). These two genes had been cloned into pUC19 by others in our lab (WHO WAS IT, pretty sure Jake and Deb?).
- Amplification
- We designed primers specifically for amplifying dhlB and dhlA, while removing a forbidden EcoRI site between them. Design with the primers allowed us to try cloning each gene by itself and also together into the shipping vector pSB1C3.
DESCRIPTION

- Cloning
- After PCR screening of the junctions between our parts and pSB1C3, we extracted the plasmids from a few that looked OK for further confirmation and submission to the iGEM HQ.
DESCRIPTION

- DOUBLE DIGEST??? PCR??? Someone decide and write something I don't have time.
We cloned the PCR fragments into pSB1C3 and transformed the ligation product. We were greatly assisted by ligating into a BBa_J04450, extracted from the linearised plasmid pSB1C3 in the Distribution Kit, which provided a neat red-white screen.
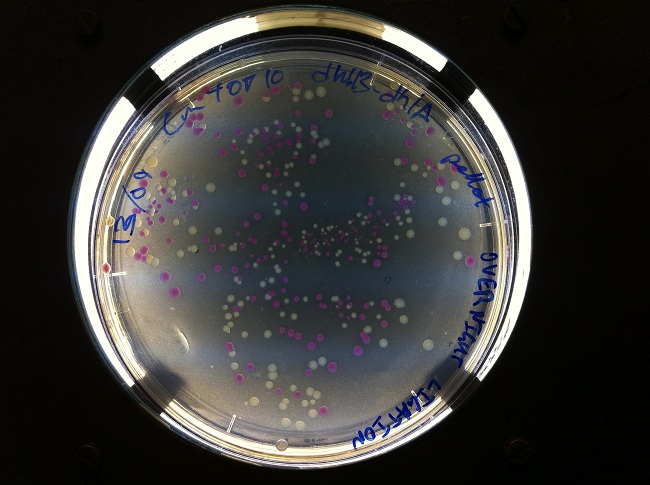
Constitutive Expression of dhlB-dhlA, Degradation of DCA and Chloroacetate
- After sending dhlB and dhlA to iGEM HQ, we began characterisation by cloning a constitutive promoter (BBa_I14033) from the Distribution Kit in front of our parts.
- Amplification
- Pcat ([http://parts.igem.org/Part:BBa_I14033 BBa_I14033]) is 38bp, but with our primers produced a 280bp fragment, 4th well from left.
- Ptet ([http://parts.igem.org/Part:BBa_R0040 BBa_R0040]) is ~50bp, but with our primers produced fragment ~300bp, 1st well from left.
- Phenotypic Assays
- After cloning (link to protocol or lab-book, digest/ligation) into a plasmid containing dhlB and dhlA, we screened for clones expressing our construct. We made screening plates (link to protocol, contained LB-agar-chloramphenicol- 10mMchloroacetate-phenol red at pH 6.8) that allowed us to pick clones that looked like they were successfully expressing one of our genes of interest.
- We showed degradation of chloroacetate and DCA by chloride assay.
With thanks to:

