 "
"
Tim Baker
From 2013.igem.org

August 8, 2013
- Checked transformation plates
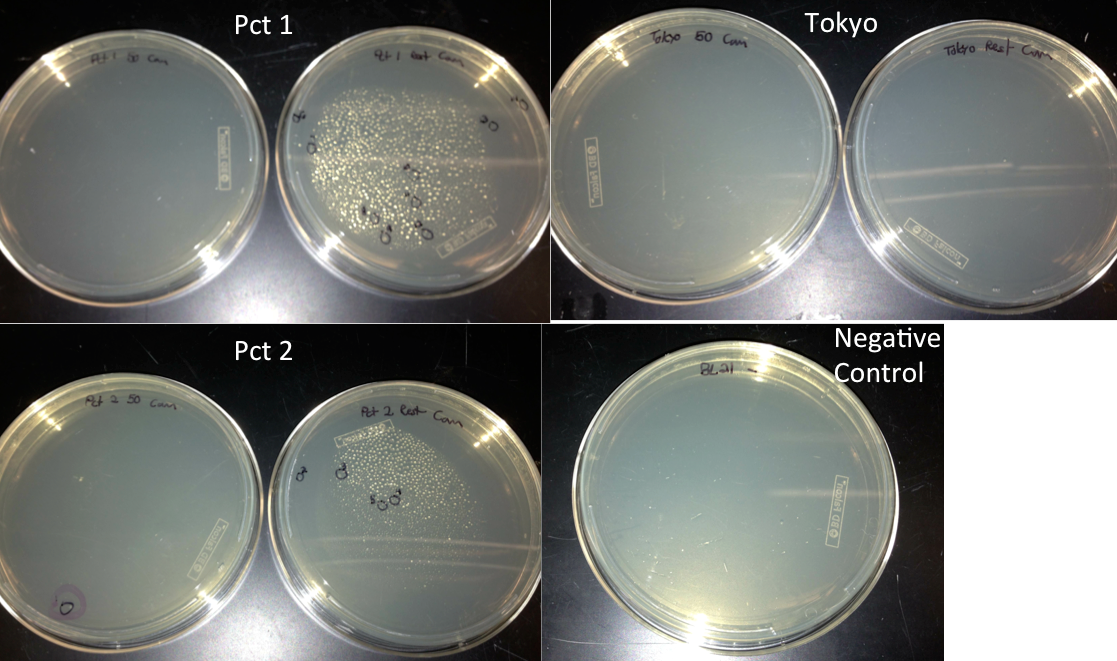
- Picked 16 colonies from Pct 1 and Pct 2 plates to be screened for the BioBrick plasmid
- Tokyo transformation did not work likely because not enough pBAD was added into the Gibson assembly to make it efficient
- Made frozen stocks of all 16 colonies to be dealt with in the Fall
- Peace out for now

August 7, 2013
- Control Experiments
- Confirming no contamination in the medium
- Made new LB min
- Confirming that BL21 doesn't have natural chloramphenicol resistance

- Confirming that the plates were made correctly with EcNR2 and XL1-Blue
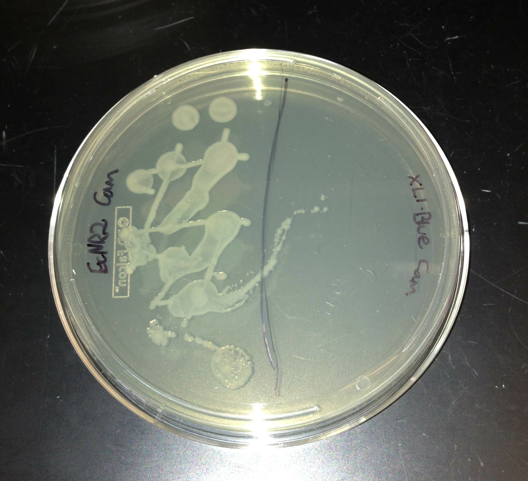
- Confirming no contamination in the medium
- All of the controls worked very well, so I assumed it was a user error and attempted the transformation again with the dsDNA from the three gibson assemblies


August 6, 2013
- The plates all showed colonies, including the negative control, which was very unusual (No results shown)
- Ran a few positive and negative controls to confirm that the BL21 strain did not have chloramphenicol resistance, that the plates were working, and that there was no external contamination
- Made new LB min to avoid contamination problems
- Made cultures of BL21 with and without chloramphenicol at the desired concentration with the new LB
- Plated EcNR2, which has chloramphenicol resistance, and XL1-Blue, which doesn't have chloramphenicol resistance
August 5, 2013
- Checked the plates from the gibson assemblies
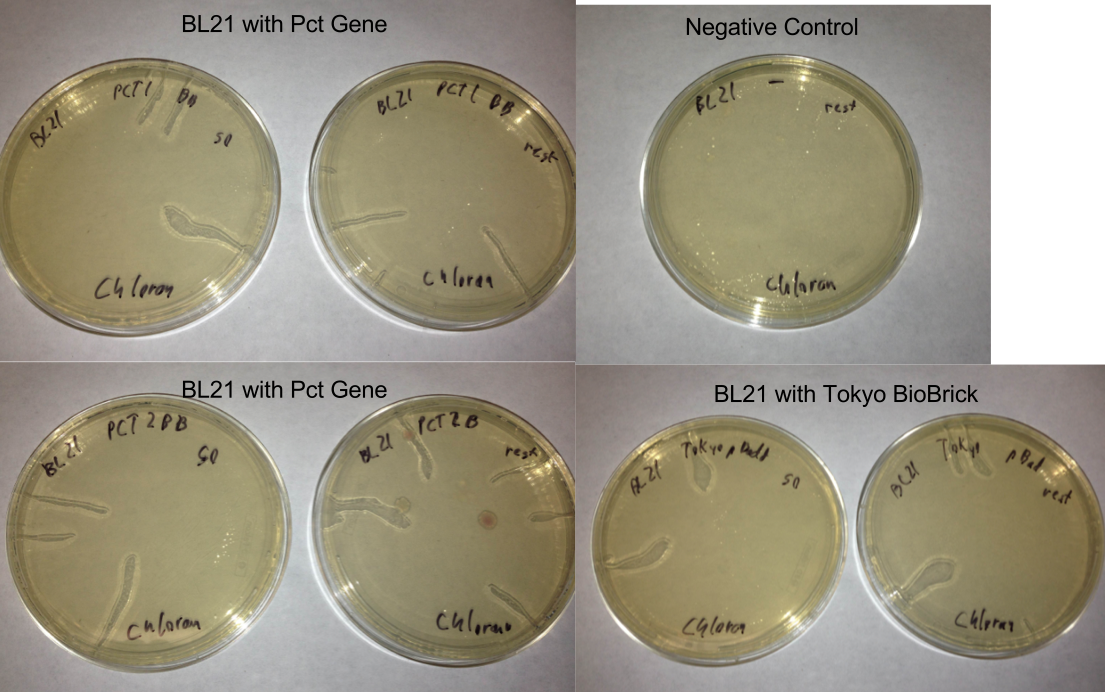
- No colonies except for a few contaminates but the plates did not look like they were made very well
- Started cultures of BL21 to redo transformation with different plates
- Made new Chloramphenicol plates
- Made up a new stock of Chloramphenicol from the solid and dissolved 20 mg/mL in 10 mL of ethanol
- Made 500 mL of LB min via the standard protocol for making medium
- Let cool down at 50 degrees Celsius then added 500 uL of the 1000x solution
- Poured 20 plates and let cool
- Ran a transformation with the new plates and the same washed gibson assembly DNA

August 2, 2013
- Putting the plasmids together and transforming
- Ran the PCR of the Pct gene on a gel and gel purified the sample to find out the nucleic acid concentration
- Using this concentration and that of the given plasmid backbone PSB1C3 set up a gibson assembly
- The Gibson assemblies of the Pct gene + PSB1C3 and of the pBAD promoter + the Tokyo part were then drop dialyzed to reduce salt concentration
- Diluted 3 uL of the washed gibson assembly into 47 uL of nuclease free water and used this as the dsDNA for a transformation
- Transformed the 2 Pct gibson assemblies and the Tokyo assembly into BL21
August 1, 2013
- Creating our own BioBrick
- The primers that we initially designed to put the two genes on a BioBrick were no longer necessary because the first gene (PHA) had 8 of the restriction sites within the genes
- To fix this, we decided to amplify only the second gene onto a BioBrick (Pct) and prove that when induced, the logic system with Pct increases PLA production
- Designed primers to amplify the Pct gene from our total gene construct with homology to the linearized PSB1C3 backbone
- Designed two sets of primers with difference homology lengths so that it would work
- Ran a PCR on the two samples in preparation for the gibson assembly
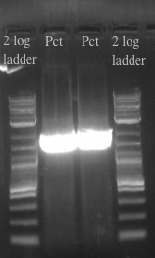

July 31, 2013
- Improving an existing BioBrick:
- The Tokyo BioBrick has been a very helpful control, but because it is expressed constitutively, its very difficult to compare to our inducible promoter
- Planning to replace the promoter with pBAD (an inducible promoter used for our PHA synthase)
- Designed primers to amplify pBAD from our gene construct with homology to the Tokyo BioBrick part and the PSB1C3 Backbone
- pBAD should be 320 bases
- Designed primers to amplify the Tokyo BioBrick part and the PSB1C3 backbone without the constitutive promoter with homology to pBAD
- Tokyo Biobrick should be 6.4 kb
- PCR and gel purified both fragments in preparation for a gibson assembly
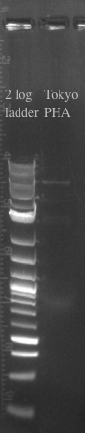
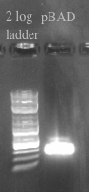
- Ran the Gibson assembly overnight


July 30, 2013
- Checked the plates from the double stranded recombination and there were no colonies on any of the plates (Results not shown)
- Not sure why this is happening but it might just be that the efficiency is very low and we are unlucky
- Ran another double stranded recombination in duplicate
- Allowed for 2 hour recovery and 15 hour recovery before plating to see if that would have any affect
July 29, 2013
- Confirmed the size of the Gene + kanR Construct via PCR
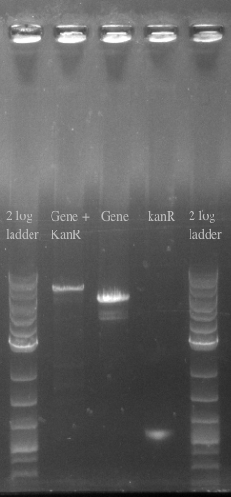
- Ran another double stranded recombination with more DNA (200 ng total) and changed the recovery time to 1.5 hours
- Re-ran because the cells that the first double stranded recombination were likely dead from being left in the shaker for too long
- Plated after 1.5 hours on Kanamycin plates

July 26, 2013
- Re-made the template from the 8 cultures of cells and then PCR amplified with primers 49 and 125 which have homology to 21B
- Ran the PCR for all of the colonies to confirm the presence of the gene
- Results:
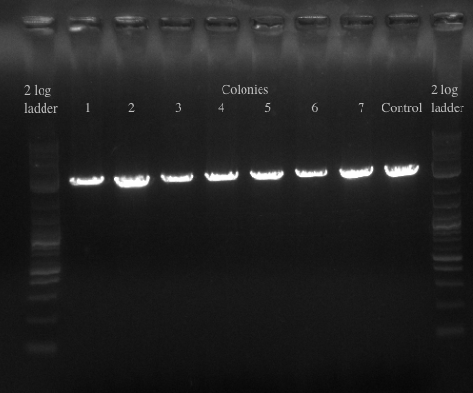
- All of the colonies showed that they had tolC at 21B
- Results:

July 25, 2013
- Checked the double stranded Recombination plates from yesterday
- EcNR1... plates showed 7 colonies on the positive (rest), 1 colony on the 50 uL, and 0 colonies on the negative control
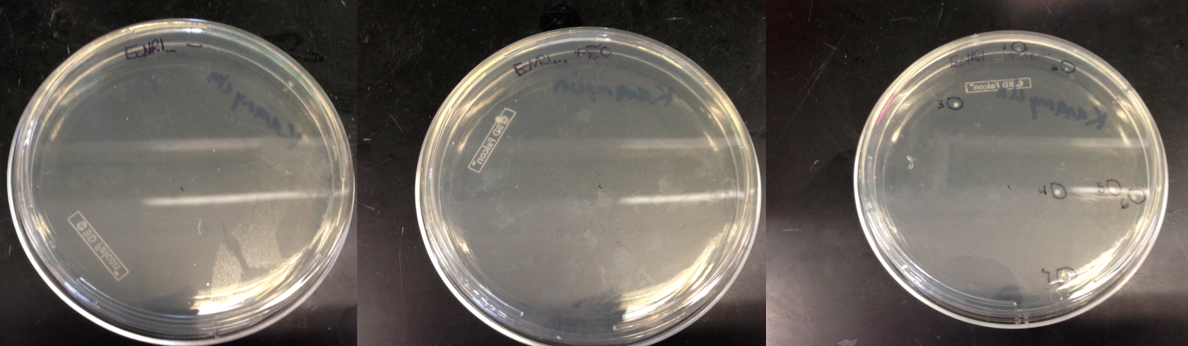
- A3 4/4 showed no colonies after growth overnight on any of the plates, so they were put back in the incubator
- EcNR1... plates showed 7 colonies on the positive (rest), 1 colony on the 50 uL, and 0 colonies on the negative control
- Picked the 7 colonies from the EcNR1 positive plate into 3 mL of LB min medium
- Let grow for a few hours, then used them as a template to screen at 21B
- Will either get a ~5.5 kb fragment (PHA/Pct + kanR) if it worked or a ~2 kb fragment (tolC) if it didn't
- Results:
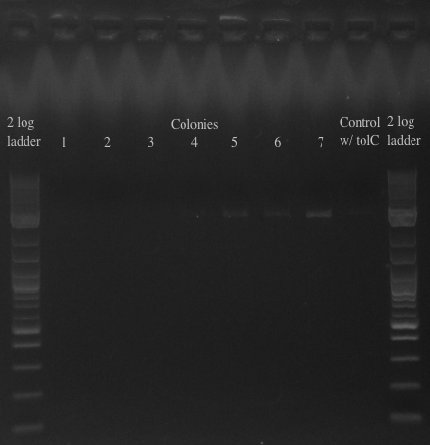
- All of the bands were not very clear, probably a result of various factors, including using a different polymerase, mixing in the primers with the mastermix, not letting the cells grow up for long enough, and the gel comb set at an unusual angle. Will redo tomorrow


July 24, 2013
- Plated the cells on Kanamycin plates
- 3 different plates for each strain that was used in the recombination
- Negative control with water as the DNA
- 50 uL of the 1 mL culture after ~15 hours of growth with 100 ng of DNA in 50 uL of water
- Spun down the cells after ~15 hours of growth from the rest of the culture and resuspended in 50 uL of water with 100 ng of DNA in 50 uL of water
- Plan on screening the 21B sight with primers tomorrow
July 23, 2013
- Grew up strains of EcNR1 mutS zeo tolC 21B and A3 4/4 which Andrew finished getting all of the knockouts on
- Strains took an unusually long time to reach midlog
- Induced cells at 42 degrees celsius and continued with the double stranded recombination protocol that Natalie provided
- Let the cells recover overnight because they have not been growing very efficiently
July 22, 2013
- Updated the Weekly Meeting presentation for the week of 7/22
- Meeting went well, Dellaporta suggested running a gradient PCR for the PHA/Pct + kanR fragment
- Checked the EcNR1... cells that were transformed with PHA/Pct on PZE21
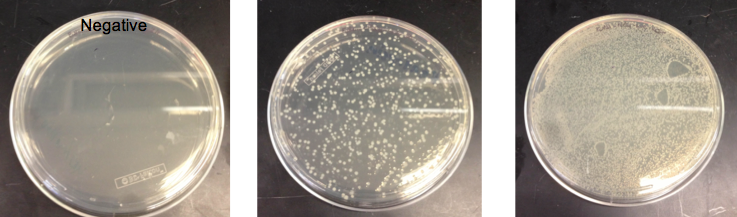
- Picked a single, medium sized, colony and started a culture for frozen stocks
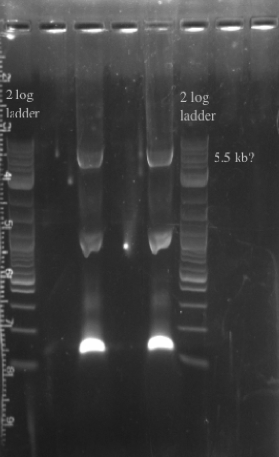
- Ran a gradient PCR on the PHA/Pct + kanR fragment as suggested by Dellaporta: Temperature range was 72-65
File:Screen Shot 2013-07-24 at 12.32.31 PM.png- The Gel shows clear bands all the way across at 5.5 kb, not sure what the other bands could be considering I used only primers 49 and 125
- Gel purified all of the bands at 5.5 kb and nano-dropped to get a concentration of 66.3 ng/uL

July 19, 2013
- Creating and transforming a plasmid with PHA/Pct
- PCR amplified PHA/Pct and PZE21 with homology to one another
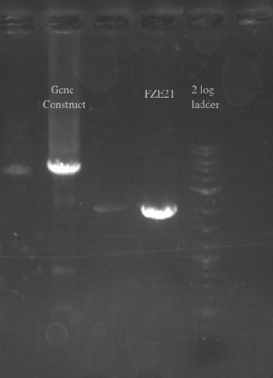
- Gibson assembled
- Performed "Drop Dialysis"
- Technique Ryan showed me to reduce salt concentration before transformations
- Transformed into EcNR1 mutS zeo tolC 21B strain
- Let grow on Kanamycin plates over the weekend
- PCR amplified PHA/Pct and PZE21 with homology to one another
- Positive Selection with kanR
- PCR amplified PHA/Pct and kanR with homology to one another
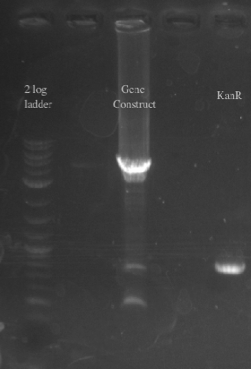
- Gibson assembled the two fragments together
- PCR amplified the Gibson assembly to increase the concentration for double stranded recombination
- Not sure whether the band is at 5.5 kb or not, will probably have to redo the PCR for better clarity
- Gel purified and nano-dropped to get a concentration of ~20 ng/uL
- PCR amplified PHA/Pct and kanR with homology to one another



July 18, 2013
- Ordered and received primers to amplify BB29 and BB30 without homology and then the PZE21 backbone with more homology
- Used BB29 and BB30 cells from frozen stocks as template DNA
- Primers 138-145
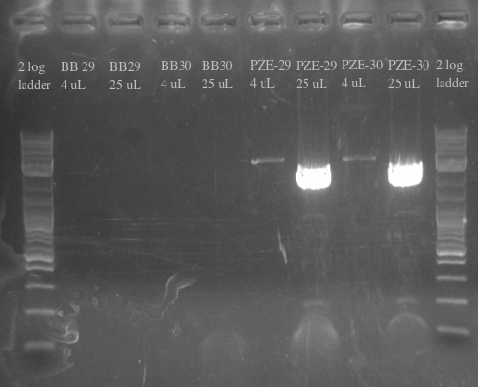
- Repeated failure to amplify the biobricks fragments suggests that there is an error in either the sequence or the shipment itself

July 17, 2013
- Ran another PCR on the Biobricks fragments and backbone to see if I could resolve the fragments using a different DNA template
- Used cells grown up overnight as the template for BB29 and BB30
- Results:
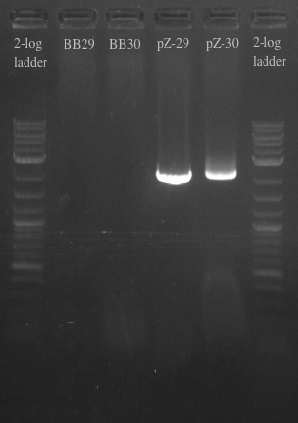
- Gel failed again pointing to something wrong with the primers
- Checked primers and re-ordered a set of primers to just amplify the BB fragments as well as amplify the backbone with longer homology for the gibson assembly
- Gel failed again pointing to something wrong with the primers

July 16, 2013
- Ran a gibson assembly and re-ran the PCR for the Biobricks fragments and the backbone on the same gel
- Results:
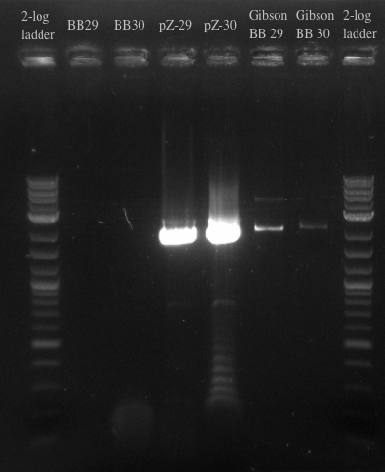
- Results:
- Results indicate that the Gibson for BB29 may have worked but there is likely something wrong with the primers for the BioBricks because the band at 900 did not show up

July 15, 2013
- Weekly meeting presentation with Farren went well: advised to forget the positive control after unpromising results
- Diluted and created working stocks of the primers for the addition of the Biobrick fragments into the expression plasmid
- Trying to create two plasmids
- Expression plasmid with PLtet-O promoter and gfp + hlya tag + terminator
- Expression plasmid with PLtet-O promoter and phasin + hlya tag + terminator
- Ran PCR gel purification to amplify the Biobricks fragments and the expression vector backbone with homology for Gibson assembly
- Nanodropped
July 10, 2013
- Continued to design sequence primers for the RBS modifications and for Knockouts
- Emailed Ken Nelson and set up a time to learn how to use the FACS Verse
- Ran Positive control on the Pseudomonas and [EcNR1 ^mutS zeo] strains grown in Nile Red according to the following procedure:
- Back-dilute 2 overnight cultures of Pseudomonas and [EcNR1 ^mutS zeo] at concentrations of 1/100 and 1/1000 in 3mL of BE and LB min medium respectively
- Add Nile Red to the positive control samples at a concentration of 0.5 ug/mL
- Allow samples to grow in shaking incubator at 34 degrees celsius for 3-4 hours
- Spin down 1mL of cells in the microcentrifuge and wash and resuspend the pellets in PBS
- Test cells on fluorescence plate reader standardized to a PBS solution
- Results: (Average of 3 trials)
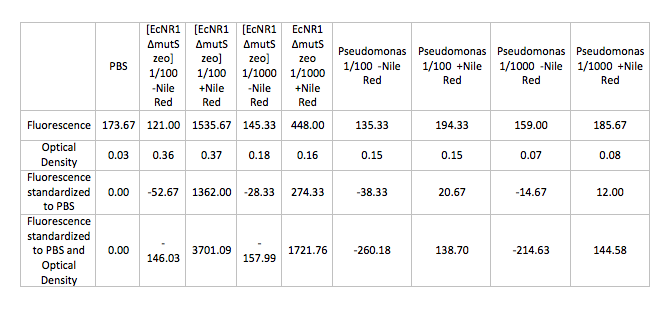

July 9, 2013
- Received strain of Pseudomonas, created 3 cultures with Beef Extract Medium and put them in shaking incubator
- 1 culture with the entirety of the concentrated pellet in 6mL of BE Medium, 2 cultures with 100 uL of the concentrated culture in 5 mL of medium
- Plated 2 cultures (concentrate and dilute) and put them in the 32 degree incubator
- Designed an experiment as a positive control for the effectiveness of the type 1 export system
- Designed Sequence Primers for RBS Modifications and for Knockouts

July 8, 2013
- Created 500mL of the 3 Nutrient Broth (Beef Extract Medium)
- 1.5g Meat Extract + 2.5g Peptone
- Created 500mL of the 3 Nutrient Agar (Beef Extract Agar) and poured 24 plates
- 1.5g Meat Extract + 2.5g Peptone + 7.5g Bacteriological Agar
- Designed experiment to test whether nile red can effectively stain Pseudomonas cells producing PHAs
- Test 4 groups: E. Coli cells washed with PBS grown with and without nile red and Pseudomonas cells washed with PBS grown with and without nile red (all standardized to PBS)
June 26, 2013

June 18, 2013
- Metabolic Engineering Graphic

June 14, 2013
- Researched metabolic engineering to increase PLA

- Took pictures of the second cycle of MAGE practice



{kind=link}
{kind=link}
June 12, 2013
- Positive MAGE Plate

- Replated negative control MAGE colonies and they worked (initial plates went bad, due to light exposure)
