 "
"
Team:Imperial College/Modelling PLAdeg
From 2013.igem.org





Contents |
Polyhydroxybutyrate (P(3HB)) Synthesis Module
Metabolic modelling
Introduction of the metabolic model
The metabolic model predicts the interaction of our pathway with endogenous pathways. In synthetic biology, an ideal system is fully orthogonal. However, if the synthetic pathway uses some metabolites from the metabolism pathways, the metabolic fluxes (the rate of conversion between metabolites) will be affected. If the cell is overloaded by losing too much metabolites, it would either reject the synthetic pathway or burn out. As for the optimization of PHB production in our MAPLE system, metabolic analysis need to be carried out to estimate the effects on the cell metabolism from increases in the PHB production.
Methods
We altered the metabolic model by Angela Dixon in her paper “Predictive Mathematical Model for Polyhydroxybutyrate Synthesis in Escherichia coli”. [ref] The metabolic model is built in Simbiology, which is an extension in MATLAB especially for biological system analysis. The model consists two metabolic pathways: Glycolysis and TCA cycle. The reactions in the metabolic pathways are defined by ordinary differential equations and kinetic parameters. The model also contains a synthetic pathway that produce P(3HB) from acetyl-coA. The pathway consists three enzymes which are PhaA, PhaB and PhaC. The reasons we chose this model:
1. The model is based on ODEs instead of FBA (flux balance analysis) method, which is a common methods of metabolic analysis. However, FBA cannot determine the real time concentration of the metabolites whereas it can be done by models of ODEs.
2. Only single gene deletion or addition can be performed in FBA. More than that, the metabolic model here can perform modification on any single or multiple genes and reactions through Simbiology.
3. The FBA model is static, it cannot analyze the dynamic interactions between our synthetic pathways with the metabolic pathways. As both our P(3HB) synthesis model and the metabolic model are in the same platform. It was really easy to integrate our pathway into the metabolic model.
In our project, we have a different pathway:
1. We use 3HB as one of the input.
2. Existence of ato system in the E.coli MG1655 strain.
3. We removed phaA in our case because we use 3HB as the major source to produce P(3HB) to avoid too much acetyl-coA be taken from the metabolic pathways.
Therefore, we substitute the original pathway with our synthetic pathway. Moreover, we added our gene expression models for the enzymes into the metabolic model as well. In terms of the consistency of the model, we changed the units of our synthetic pathway into micro molar and seconds.
Here is the combination of our model with the metabolic model
[[]]
We also carried out a parameter estimation of the initial concentrations of several species in our synthetic pathway. The reason is we have single-cell scaled magnitude for them originally (fM level), but there are much higher magnitudes in any of the species in the metabolic pathways (mM level). Therefore, we scaled up the pathway in order to maintain the consistency of the model. As for the metabolites which are involved in our pathway, we cloned the blocks of them from the metabolic pathway in Simbiology and deliberately put them into our pathway. That means our pathway dynamically interact with the metabolic pathways.
| enzyme | source organism | biobrick | reference |
|---|---|---|---|
| Proteinase K | Engyodontium album | [http://parts.igem.org/Part:BBa_K1149002 BBa_K1149002] | [http://www.ncbi.nlm.nih.gov/nuccore/X14689.1] |
The overall PLA degradation model is shown as below:

There are two compartments which represents cells and the culture from left to right. The "cell" compartment contains the gene expression module whereas the "culture" compartment contains the degradation module. The "secretion" block that connects two compartments is the secretion module.
Parameters and assumptions
Gene expression module of Proteinase K
| Parameter | Description | Value | Units | Sources | Assumptions |
|---|---|---|---|---|---|
| β | maximum rate of transcription | 0.032 | mM/min | Please see derivation 1 below. | Please see derivation 1 below. |
| K | Activation coefficient | 0.0031 | mM | [http://parts.igem.org/Part:BBa_K206000:Characterization] | Taking the "switch point" as the activation coefficient |
| dmRNA | mRNA degradation rate | 0.10 | 1/min | [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC420366/pdf/07X.pdf] | Taking the value of mRNA half-life in E.coli strain MG1655 as 6.8min. rate = ln2/half-life = ln2/6.8 = 0.10 |
| dprotein | Protein degradation rate | 0.050 | 1/min | [http://jb.asm.org/content/189/23/8746.full] | There is no active degradation pathway and that dilution is the dominant way by which the protein level decreases in a cell. Rate = 1/doubling time, where doubling time = 20min. Assuming steady-state growth in LB broth as presented in paper. |
| k2 | Protein production rate (Proteinase K) | 4.7 | 1/min | Please see derivation 2 below. | Please see derivation 2 below. |
| [Arabinose] | Concentration of arabinose | Initial: 0.008 | mM | ||
| [mRNA] | Concentration of mRNA | - | mM | - | - |
| [Proteinase K] | Concentration of Proteinase K | - | mM | - | - |
1.Derivation of the maximal expression rate,β
- Average molecular weight (Mw) of a base pair = 660g/mol[http://www.geneinfinity.org/sp/sp_dnaprop.html][http://www.lifetechnologies.com/uk/en/home/references/ambion-tech-support/rna-tools-and-calculators/dna-and-rna-molecular-weights-and-conversions.html]
- Average mass of a base pair = 660g/mol x 1.66x10-24 = 1.1x10-21g
- Volume of an E.coli cell = 1µm3[http://kirschner.med.harvard.edu/files/bionumbers/fundamentalBioNumbersHandout.pdf] = 1x10-15L
- ∴Mass concentration =
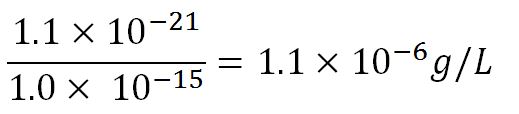
- ∴Molar concentration of 1 base pair in the volume of E.coli =
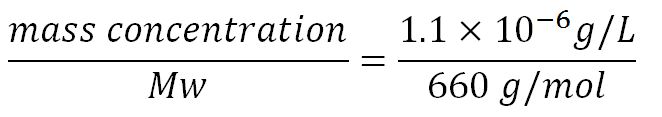 = 1.66x10-6 mM
= 1.66x10-6 mM
- ∴Mass concentration =
- BioBrick assembly plasmid pSB1C3 is a high copy number plasmid (100-300 copies per cell)[http://parts.igem.org/Part:pSB1C3?title=Part:pSB1C3]
- assume 200 copies per cell
- ∴ concentration of the gene per cell = N x 200 x 1.66x10-6mM, where N = number of base pairs
- ∴ concentration of the gene proteinase k (N = 768) in the volume of an E.coli cell is = 0.25mM
- Transcription rate in E.coli = 80bp/s[http://kirschner.med.harvard.edu/files/bionumbers/fundamentalBioNumbersHandout.pdf] = 80 x 1.66x10-6mM/s = 80 x 1.66x10-6 x 60mM/min = 7.97x10-3mM/min
- ∴ Rate of mRNA_Proteinase K production under the control of pBAD = 7.97x10-3 ÷ 0.25 = 0.032/min

- Average molecular weight(Mw) of an amino acid(aa)= 110g/mol[http://www.genscript.com/conversion.html][http://www.promega.com/~/media/Files/Resources/Technical%20References/Amino%20Acid%20Abbreviations%20and%20Molecular%20Weights.pdf]
- Average mass of an amino acid = 110g/mol x 1.66x10-24=1.83x10-22g/L
- ∴Mass concentration of one aa in the volume of an E.coli =
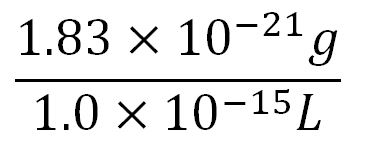 = 1.83x10-6g/L
= 1.83x10-6g/L
- ∴Molar concentration of one aa =
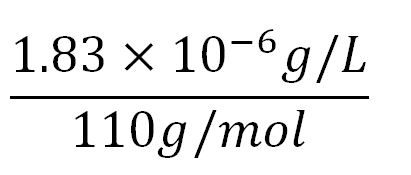 = 1.66x10-5mM
= 1.66x10-5mM
- ∴Mass concentration of one aa in the volume of an E.coli =
- Translation rate = 20aa/s = (20 x 1.66x10-5 x 60)mM/min = 0.020mM/min
- proteinase K comprises of 256aa[http://www.uniprot.org/uniprot/Q2PEN2&format=html]
- ∴concentration of proteinase k's aa in the volume of an E.coli = 1.66x10-5mM x 256 = 4.25x10-3mM
- ∴ Rate of protein production = 0.020 ÷ 4.25x10-3 = 4.7/min


Degradation
The reaction equation of the PLA degradation is:
[Polylactic acid]+[Proteinase K]= 660 [lactic acid] + [Proteinase K]
Assumptions:
We assumed 1 mole of polylactic acid can produce 660 moles of lactic acid.
The molecular weight of a single polylactic acid monomer is 90 g/mol [http://www.chemspider.com/Chemical-Structure.592.html]whereas the molecular weight of the solid polylactic acid is around 59500 g/mol.[http://www.sciencedirect.com/science/article/pii/S0014305711003582]Therefore, the short-chain polylactic acid consists approximately 660 monomers. 660 molecules of lactic acid will be produced by degrading one chain of polymer.
We also assumed a simple Michaelis-Menten mechanism for proteinase K

The parameters for the kinetic equations are:
| Parameter | Description | Value | Units | Sources |
|---|---|---|---|---|
| Km | Michaelis constant | 0.032 | mM | - |
| Vmax | Maximum velocity | 2.472 | mM/min | - |
| Proteinase K | Concentration of the enzyme in assays | 30 | mg/L | - |
| Kcat | turnover number | 2348.4 | 1/min | See derivation below |
| Mw | Molecular weight of proteinase K | 28.5 | KDa | [http://www.sigmaaldrich.com/etc/medialib/docs/Sigma/Datasheet/2/p4850dat.Par.0001.File.tmp/p4850dat.pdf] |
Derivation:Turnover number (Kcat) = Vmax*Mw/[proteinase K] = 2348.4
The efficiency of Secretion is assumed to be 90% secretion over 2 hours.[http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1251600/] The rate of secretion in the model is therefore:
rate of secretion = 0.9[concentration of Proteinase K]/120 (mM/mins)
Simulations and Results

{kind=link}