 "
"
Team:British Columbia/Modeling
From 2013.igem.org
Joelkumlin (Talk | contribs) (→Simulating virus-mediated population control) |
(→Extending to Co-culture) |
||
| Line 190: | Line 190: | ||
[[File:Theoretical_production.png|600px]] | [[File:Theoretical_production.png|600px]] | ||
</center> | </center> | ||
| - | <h4 align = "center"> Figure 5 - Theoretical product formations based on co-culture dynamics | + | <h4 align = "center"> Figure 5 - Theoretical product formations based on co-culture dynamics.</h4> |
==Probabilistic Model== | ==Probabilistic Model== | ||
Revision as of 01:54, 29 October 2013
iGEM Home


Population Dynamics Modeling
Mixed cultures are often used in bioprocessing, where the quality of the final product requires optimal strain balance. We realized that mixed cultures harbouring different CRISPR assemblies could be modulated by applying selective pressure against targeted strains via phage addition. This provides a novel opportunity for tuning bacterial consortia, directly applicable to industrial bioprocesses such as yogurt production that rely on mixed culture fermentation. Here, we develop deterministic and probabilistic models to provide a predictive framework for optimizing strain balance within mixed populations.
We then extend this model to allow for optimization of product formation, such as flavouring of yogurt.
Our main objectives were as follows:
- Predict the growth of recombinant E. coli cultures under phage predation.
- Predict vanillin and cinnamaldehyde production based on initial ratio of virus to bacteria, (ie. multiplicity of infection; MOI).
To help conceptualize our model, we first generated the schematic shown in Figure 1. For simplicity, we considered two E. coli strains, each harbouring two plasmids: a flavouring plasmid and a CRISPR (or "immunity") plasmid. Here, the flavouring plasmid is responsible for the production of either cinnamaldehyde or vanillin and the CRISPR plasmid contains unique spacer elements, granting both strains immunity against "environmental phage" that may contaminate the bioreactor. Additionally, one strain would be immunized against the "control phage", where the second strain would be susceptible to viral infection. This would allow us to tune the susceptible population in order to achieve our desired flavouring proportions, based on the amount of control phage added.
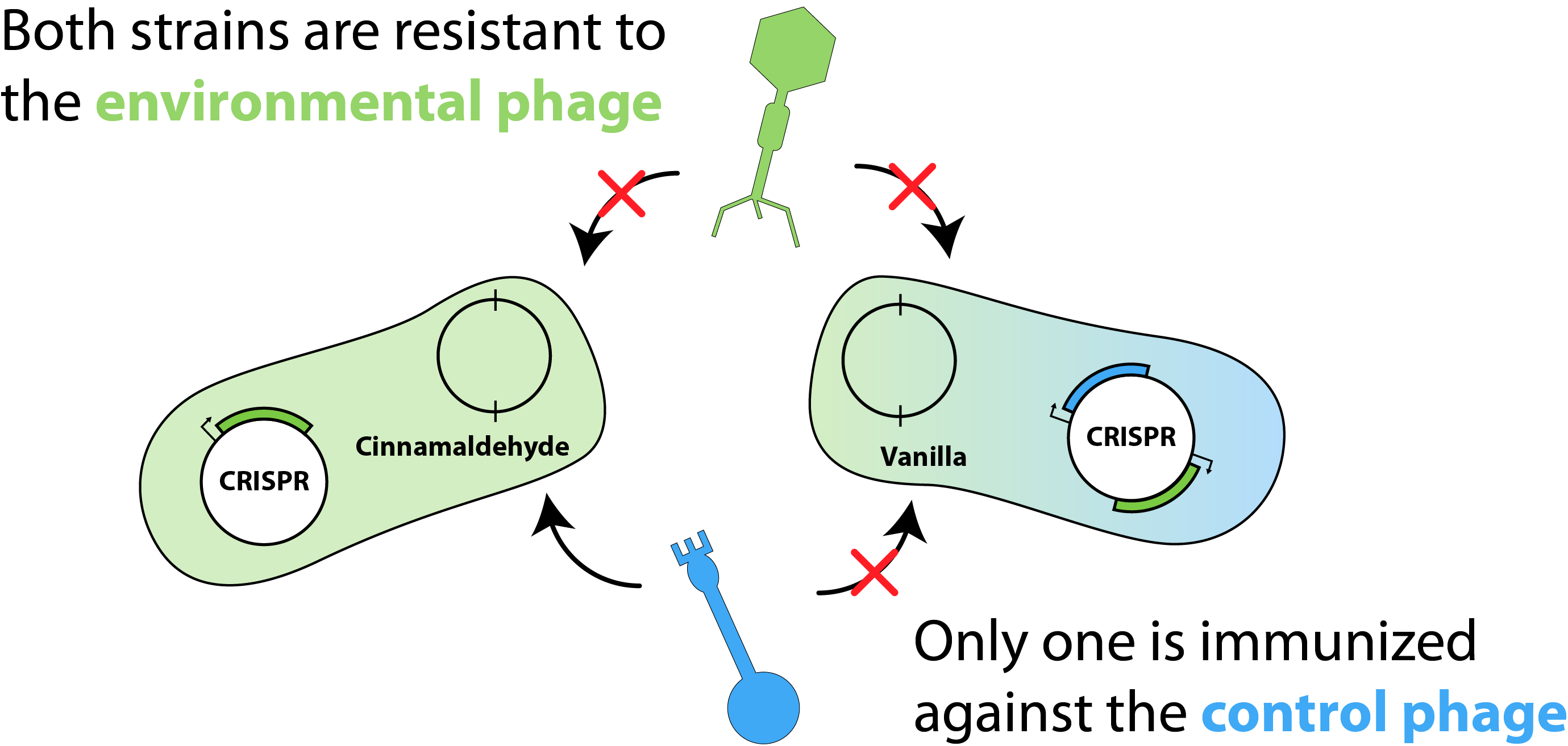
Figure 1 - Model Schematic.
Modeling bacteria growth under phage predation
A deterministic model based on Monod kinetics [1] was used to describe bacterial growth. Here, the Poisson distribution was incorporated to model the infected populations based on a starting phage-to-bacteria ratio, or MOI. We trained the model on a set of experimental growth curves and validated it against a separate set. Yield terms were added to account for the production of two different flavours: cinnamaldehyde and vanillin. Next, we extended our basic growth model to include bacterial co-cultures. Using this approach, we demonstrate that by using different virus inoculums, it is possible to achieve different final amounts of compounds from a mixed culture.
Simulating virus-mediated population control
To simulate the behaviour of cells growing in mixed populations, we computationally modelled the co-cultures of two flavouring product strains using the cell programming language "gro" [2]. The model considers two strains of bacteria on a two-dimensional surface under phage predation. One of the strains is immune to infection, whereas the other is susceptible. Here, we use gro as a framework for visualizing the interactions between phage and mixed bacterial populations with and without the CRISPR assembly. From our simulations, we see that phage-mediated control of bacterial populations is indeed a suitable method of tuning bacterial consortia for optimization of product production.
Model Formulation
Assumptions
- Bacteria is grown in a batch culture and follows the Monod growth equation.
- Bacteria with the CRISPR are nearly 100% immune to the specific phage infection.
- Bacteria without CRISPR system are completely susceptible to phage infection.
- Yield coefficients and phage attachment coefficients are constants.
- Bacterial phages do not decrease significantly in number during the course of an experiment.
- All considered bacterial phages are lytic.
Deterministic Model
Bacteria Growth
We begin with a populations balance for a given strain $X$, \begin{align} \frac{dX}{dt} = \frac{dX_i}{dt} + \frac{dX_u}{dt} \end{align} The infected bacteria is represented by $X_i$, uninfected bacteria by $X_u$ and $X = X_i + X_u$. We use the Monod equation to model the substrate limited bacterial growth. \begin{align} \mu_{X} = \mu_{max, X} \frac{S}{K_{S, X} + S} \end{align}
Where $\mu_{X}$ is the bacterial growth rate, $\mu_{max, X}$ is the maximum bacterial growth rate, $S$ is the growth limiting substrate and $K_s$ is the amount of substrate remaining when the growth rate is half of maximum. Due to environmental factors, some bacteria will die; the death rate ($k_d$) is assumed to be directly proportional to the bacteria population. A lag phase is modelled by multiplying a dampening term to the growth rate, namely $(1 - e^{-\alpha t})$, where $\alpha$ is defined as a proportionality constant. The slowing of growth from exponential to stationary is modelled with yet another dampening term to be multiplied to the growth: $exp\big[{-\big(\frac{X}{X_c}\big)^m\big]}$. This dampening term is related to the quorom sensing in bacteria, once the bacteria reach some characteristic concentration ($X_c$) the bacteria begin to slow down growth. Thus the uninfected cell growth is described by: \begin{align} \frac{dX_u}{dt} = \big( \mu_{X, u}e^{-\big(\frac{X}{X_c}\big)^m} - k_{d_{X,u}} \big) X_u\big(1 - e^{-\alpha t}\big) \end{align} For simplicity, in this model, $m$, $X_c$, and $\alpha$ are all to be empirically determined and assumed constant. In reality, each constant is dependent on many variables, for example: temperature, pressure, [substrate], environmental conditions, bacterial strain, and more. Similarly for infected cells: \begin{align} \frac{dX_i}{dt} = \big( \mu_{X, i}e^{-\big(\frac{X}{X_c}\big)^m} - k_{d_{X,i}} \big) X_i\big(1 - e^{-\alpha t}\big) - \delta \end{align}
However, a major difference is that the decay rate for the infected cells is dependent on the latency time of the virus and thus we introduce $\delta$. The function $\delta$ is defined as follows,
Here $N(\tau)$ is a normal distribution over some time $\tau$, in other words, it is expected that the infected population will lyse according to a normal distribution over the domain $(-\tau, \tau)$.
Viral Growth
It is now necessary to consider the phage population $V$ as it directly affects the amount of bacteria that are infected at any given time. To describe the phage population it is necessary to consider how many phages are released per cell lysis, this number is referred to as the burst size $(\beta)$. \begin{align} V = V_0 + X_i \beta ^{\frac{t}{LT}} \end{align} Where $V_0$ is the initial phage populations and $LT$ is the latency time (they time between infection and lysis). However, since lab techniques are tricky to measure the exact phage at various time points as well as the exact amount of infected bacteria, it is desired to simplify the model. It is reasonable to use a stepwise function that describes the amount of free phage present. Also, since the burst size is large, on the order of $100$ [3], we see that the loss of current phage due to infection of cells is small (roughly 1%), thus to simplify computations we consider this to be negligible. With these deductions we arrive at the following: \begin{align} V = V_0 + E(X_i)\beta^{\big[\frac{t}{LT}\big]} \end{align} Where $E(X_i)$ is the expected infected population and $\big[\frac{t}{LT}\big]$ is the largest integer equal or less than $\frac{t}{LT}$. To find the expected infected population, the Poisson distribution is used based on the MOI. However, there is an efficiency of viral attachment $\epsilon \in (0, 1)$ (in fact the efficiency has been found to be a function of certain proteins) [4,5]. \begin{align} MOI = \frac{V}{X} \end{align}
\begin{align} E(X_i) = \epsilon X(1 - e^{-MOI}) \end{align}
Substrate Utilization
The bacteria will need to use a substrate, and the depletion of this substrate is proportional to the growth of the uninfected bacteria and the amount of bacteria. Although the infected bacteria do not multiply when infected, the infected cells may use up substrate to gain the energy needed to replicate the phage. Although it may be the case that the bacteria simply recycle intracellular material, a substrate utilization term ($\gamma$) for the infected cells is utilized (if the bacteria do in fact recycle intracellular material, it follows that this value will be 0). Moreover, when cells lyse, the cell materials can be metabolized by other bacteria and will add to the amount of nutrients available. Thus, the equation for substrate utilization rate is:
\begin{align} \frac{dS}{dt} = - \big[ \frac{\mu_u}{Y_u}X_u + \frac{\gamma}{Y_i}X_i \big] + \phi \delta \end{align} Where $Y_u$ and $Y_i$ are yield constants and $\phi$ is the average amount of substrate released per infected cell during lysis. For clarity, $\delta$ is as described above.
Model Validation
Although, only limited amounts of growth curves have been able to be analyzed, the initial quantitative results appear promising and the qualitative results have been found to contain information whether or not the quantitative results align. The qualitative curve were especially good at predicting culture collapse (keep in mind that OD measurements will contain "bacterial bones" which will artificially inflate the concentration of bacteria after lysis).

Figure 2 - Bacteria Growth curves from lag phase to approaching stationary phase. The model was optimized on another set of data and was tested for validity on this set.

Figure 3 - Bacteria Growth under phage predation. In models where the quantitative values differ, there is still qualitative information that can be extracted.

Figure 4 - A better fit for bacteria Growth under phage predation. Also included, the predicted infected and uninfected populations.
Product Formation
For a given uninfected bacteria $X_u$ that can produce the product $P$, we will first expect the production rate to be first order equation: \begin{align} \frac{d[P]}{dt} = \frac{\mu_{X_u}}{Y_P}X_u \end{align} Where $Y_P$ is the yield coefficient of the product. For infected bacteria we expect the product formation rate to be negligibly small.
Extending to Co-culture
The end goal of our project is to tune product formation of two or more compounds by adding phage to a batch reactor. Our extension to co-cultures considers the situation where vanillin and cinnamaldehyde strains have both been engineered with the CRISPR immunity to an environmental phage. Additionally, the vanillin producing strain has been engineered with immunity to a control phage, whereas the cinnamaldehyde is susceptible. Since both strains contain the CRISPR system and an engineered metabolic pathway it is not entirely unreasonable to assume that growth rates are similar. Let our bacterial cultures be $X$ for cinnamaldehyde and $Z$ for vanillin. Our equations become:
Subject to initial and boundary conditions:
We use Matlab to solve this system of equations. This model expects to show how phage can be used to control product formation, but to know whether or not the model paints the right picture we must first have a qualitative understanding of what we are expecting to see. As the initial MOI of the control phage increases, the cinnamaldehyde producing strain will collapse faster than at a lower MOI. If we assume that the production rates of vanillin and cinnamaldehyde equal during growth without phage, then we expect the product formation curves to separate with increasing MOI. Figure 5 shows that our model run at two different MOI's (two orders of magnitude apart), notice that the model predicts the qualitative trends we were expecting to see.

Figure 5 - Theoretical product formations based on co-culture dynamics.
Probabilistic Model
For the numerical simulation of co-cultures we utilized the cell programming language gro, which was developed by the Klavins Lab at the University of Washington [2]. The model considers two strains of bacteria on a two-dimensional plane under attack of one virus. One strain (green) contains a specific spacer element for the "control" phage and is granted immunity, whereas the other stain (yellow) is susceptible to phage infection. In the simulation, susceptible bacteria entering regions with high phage concentrations are very likely to become infected and upon lysis increase the phage concentration of that region. Below is a timeseries illustrating the control of susceptible cells with phage addition over the course of one batch cycle. Here, the amount of viable susceptible cells remaining over time can be controlled by adjusting the starting phage-to-bacteria ratio, allowing for optimal product formation.

A simulation of the population dynamics occurring in one batch cycle is presented below:

Model Formulation: Joel Kumlin, Chris Lawson, Joe Ho
References
[1] Shuler, M.L. & Kargi, F. (2002). Bioprocess Engineering: Basic Concepts (2nd ed.). Prentice Hall, Upper Saddle River, NJ.
[2] Jang, S.S., Oishi, K.T., Egbert, R.G., & Klavins, E. (2012). Specification and simulation of multicelled behaviors. ACS Synthetic Biology 1 (8), pp 365–374.
[3] Simon, E. H., Tessman, I. (1963). Thymidine-requiring Mutants of Phage T4. PNAS, 50, 526–532.
[4] Storms, Z. J., Arsenault, E., Sauvageau, D., & Cooper, D. G. (2010). Bacteriophage adsorption efficiency and its effect on amplification. Bioprocess and Biosystems Engineering, 33(7), 823–31.
[5] Storms, Z. J., Smith, L., Sauvageau, D., & Cooper, D. G. (2012). Modeling bacteriophage attachment using adsorption efficiency. Biochemical Engineering Journal, 64, 22–29.