 "
"
Team:TU-Eindhoven/LabJournal
From 2013.igem.org
| Line 1,702: | Line 1,702: | ||
$(this).tab('show'); | $(this).tab('show'); | ||
}); | }); | ||
| - | $('#weekTabs li:eq( | + | $('#weekTabs li:eq(6) a').tab('show'); |
</script> | </script> | ||
</html> | </html> | ||
Revision as of 19:43, 26 August 2013


Introduction
Week 1
Creating agar plates
Before any real lab work could begin some supplies had to be created, including a selection of agar plates. We needed two different types of agar plates, some with ampicillin antibiotics and others with kanamycin antibiotics. To manage this we created 2 separate agar solutions, a 200mL mixture for the ampicillin plates and a 400mL mixture for the kanamycin plates. The protocol we followed for the creation of the agar solutions was as follows:
- Mix together the following amounts to create 200mL agar solution:
- 2g Peptones
- 2g NaCl
- 1g Yeast extract
- 3g Agar
- Fill the container up to 200mL with demineralised water.
- For a 400mL solution mix together:
- 4g Peptones
- 4g NaCl
- 2g Yeast extract
- 6g Agar
- Fill the container up to 400mL with demineralised water.
- Next the containers were autoclaved and allowed to cool, but not harden.
- The following steps were all performed in the vicinity of a blue flame to ensure a sterile working environment.
- Before pouring the plates the correct antibiotics were added. The concentration of the ampicillin antibiotics was 100ng/µL and that of the kanamycin antibiotics was 30ng/µL.
- The solutions were now ready to be poured into agar plates. From the solutions we made we were able to pour 8 ampicillin plates and after receiving a little extra (about 350mL) kanamycin agar solution we poured a total of 27 kanamycin plates.
- The plates were cooled on the bench and allowed to harden before being stored in a 4°C refrigerator.
Transforming the DNA
At this moment in time only one DNA vector (Protamine-1-Optimized) had arrived, but being impatient and rearing to go we decided to proceed with this one sample anyway which also allowed us a chance to get back into the rhythm of doing lab work. We needed to transform the 4µg of DNA into NB bacteria ready for plating and culturing. This all was done by completing the following steps:
- The first step was to dilute the 4µg of vector in 20µL of MilliQ water. This created a 200ng/µL solution.
- Of the previously created solution 1µL was pipetted into 199µL of MilliQ water creating a 4000 times dilution (1ng/µL).
- Of this 1ng/µL solution 1µL was pipetted into 20µL of NB bacteria. The two dilutions could now be stored in the -20°C freezer.
- The NB/Vector solution was left on ice for a short while before being heat-shocked in a water-bath of 42°C for 30 seconds.
- Now the NB bacteria were returned to ice for 2 minutes.
- After allowing the bacteria to cool 80µL of SOC solution was added. Hereafter the NB bacteria were not returned to ice. Instead they were placed in a 37°C incubator for 60 minutes.
Plating the bacteria
After incubating the bacteria for a hour they were ready to be plated so that we could create a number of cultures. This was done in the following fashion:
- The following steps were all performed in the vicinity of a blue flame to increase the sterility.
- The plate was opened and the entire bacterial solution (approx. 101µL) was pipetted onto an ampicillin agar plate.
- To ensure even culture growth the solution was spread out over the plate using a sterile spreader.
- The plate with its bacterial spread was then placed in a 37°C incubator and left there overnight to grow.
Making LB medium
Another preparational step that was performed was the creation of LB medium which we will be using a lot in the coming weeks. The protocol for the making of LB medium follows that for the creation of agar solution without the addition of the agar. So to create 1L of LB medium we mixed the following:
- 10g Peptones
- 10g NaCl
- 5g Yeast extract
- Fill the container up to 1L with demineralised water.
- The entire container was then placed in the autoclave and sterilized.
- The LB medium was allowed to cool and has been stored for later use at room temperature.
Creating small cultures
Today we continued with the Protamine-1-optimized sample. This sample had been plated onto an ampicillin agar plate and was allowed to grow overnight. The next step was to transfer cultures from this agar plate into small amounts of LB medium so we could obtain a slightly larger culture, essentially increasing the amount of viable DNA vectors we have. The transformation to LB went as follows:
- All following steps were performed in the vicinity of a blue flame increasing the sterility.
- Firstly three small falcon tubes were filled with 8mL of LB medium.
- To each of the falcon tubes 8µL of ampicillin antibiotics were added.
- Now the agar plate was opened and three free lying colonies were chosen for picking. Each of the chosen colonies was then picked by lightly scraping across it with a pipet point. Each pipet point and colony picking was then ejected into one of the falcon tubes.
- The falcon tubes were then placed into a rotating incubator set to 37°C and left there to culture overnight.
As there were no further preparational steps to perform and no other DNA vectors had arrived this concluded a rather short day in the lab.
DNA Retention from cultures
Previously (on the 25th July 2013) three small 8mL cultures had been placed in the incubator containing NB bacteria which housed the vector for the protamine-1-optimized sample. These cultures had grown overnight and it was our aim today to retain the DNA vectors from these three cultures, essentially having allowed us to multiple the amount of DNA we had available. To perform this DNA retention a miniprep protocol was followed as is described below:
- First the culture tubes were spun down in a centrifuge for 10 minutes at 3700rpm causing the bacterial cells with the DNA inside to form pellets isolating it from the growth medium.
- The supernatant that had formed above the pellets was discarded and the pellets themselves were resuspended in 250µL of P1 buffer. The tubes were gently shaken by hand until the entire pellet had resuspended.
- The suspension was then transferred by means of pipetting into a smaller (1.5mL) eppendorf tube.
- 250µL of P2 buffer was then added to the eppendorf tube and the solution was inverted by hand until it had turned a clear blue.
- Within 5 minutes of adding the P2 buffer 350µL of N3 buffer was added to the solution and the eppendorf tube was inverted again until the solution was clear again.
- Once clear the eppendorf tube was placed in the centrifuge and spun for 10 minutes at 13000rpm seperating the bacterial cells from the DNA which remained suspended in the solution. The bacterial cells formed a pellet in the bottom of the eppendorf tube. (Unfortunately the pellet had not completely formed after 10 minutes so the eppendorf tube was spun for a further minute, again at 13000rpm.)
- As soon as the centrifuge had stopped and we could see that the pellet had properly formed, the supernatant was poured off into a special QIAcolumn. The pellet could be discarded in the bio-hazard waste.
- The QIAcolumn was subsequently centrifuged for one minute at 13000rpm, during which the DNA vector bound to the column meaning the flow through could be discarded.
- 750µL of PE buffer was then added to the QIAcolumn before centrifuging it for another minute at 13000rpm. The flow through was discarded.
- To ensure that all the PE buffer had passed through the column it was then spun yet again for one minute at a speed of 13000rpm. Yet again the flow through could be discarded.
- The column itself was then placed above a sealable 1.5mL eppendorf tube so that the DNA could be contained and stored. To obtain the DNA from the column 50µL of MilliQ water was pipetted onto the very center membrane of the column after which it was spun down for one minute at 13000rpm. The flow through now sat in the 1.5mL eppendorf tube and contained the DNA vectors.
Nanodrop Test
To check how much DNA we had acquired by culturing the NB bacteria with our vector a small nanodrop test was performed. This would tell us how many ng of DNA could be found in each µL of our samples. The results have been given below:
- Tube 1 contained 145.5 ng/µL of DNA.
- Tube 2 contained 164.1 ng/µL of DNA.
- Tube 3 contained 153.9 ng/µL of DNA.
The remaining DNA was then placed in a -20°C freezer and stored for later use concluding the days lab work.
Week 2
Transforming DNA
Today a further 4 constructs arrived (Poly(Arginine-Serine), Poly(Arginine-Glycine), 1PJN1 and 1ETF) so it became possible to continue with the lab work: increasing the amount of viable DNA for these samples much like we had done for the Protamine-1-optimized sample which we had recieved a week earlier. To increase the DNA we would transfer the DNA into Nova-Blue bacteria and culture these allowing the bacteria to replicate our own vector constructs. To isolate the DNA we would then move on to miniprepping the cultures. Firstly however the would need to be transformed into the bacteria. The steps taken to do this are listed below:
- Before transformation could be started the 4µg of DNA we had recieved would need to be diluted to a 1ng/µL concentration as follows:
- The first step was to dilute the 4µg of vector in 20µL of MilliQ water. This created a 200ng/µL solution.
- Of the previously created solution 1µL was pipetted into 199µL of MilliQ water creating a 4000 times dilution (1ng/µL).
- With this 1ng/µL solution the transformation could commense:
- Of this 1ng/µL solution 1µL was pipetted into 20µL of NB bacteria. The two dilutions could now be stored in the -20°C freezer.
- The NB/Vector solution was left on ice for a short while before being heat-shocked in a water-bath of 43.6°C for 30 seconds. (This should have been 42°C but the temperature gauge was thought to be broken at this time.)
- Now the NB bacteria were returned to ice for 2 minutes.
- After allowing the bacteria to cool 80µL of SOC solution was added. Hereafter the NB bacteria were not returned to ice. Instead they were placed in a 37°C incubator for 60 minutes.
Plating the bacteria
After incubating the bacteria for a hour they were ready to be plated so that we could create a number of cultures. This was done in the following fashion:
- The following steps were all performed in the vicinity of a blue flame to increase the sterility.
- The plate was opened and the entire bacterial solution (approx. 101µL) was pipetted onto an ampicillin agar plate.
- To ensure even culture growth the solution was spread out over the plate using a sterile spreader.
- The plate with its bacterial spread was then placed in a 37°C incubator and left there overnight to grow.
Creating the Cultures
Looking ahead to the cloning we would like to perform later on and using the yields of DNA obtained from the Protamine-1-Optimized sample last week a small calculation was performed to see how much DNA we would need and therefore how many cultures would need to be created. As we were going to attempt to use gel extraction to purify the digestion products of these constructs we would need to culture approximately 20 times more DNA than that we would eventually need for the ligation. Based on the average 150ng/µL DNA yield from the Protamine-1-Optimized construct we determined we would need 12 8mL cultures per construct to obtain the correct amount of DNA. This meant that we would would be creating:
- 12 cultures of Poly(Arginine-Glycine).
- 12 cultures of Poly(Arginine-Serine).
- 12 cultures of 1PJN1.
- 12 cultures of 1ETF.
- 9 cultures of Protamine-1-Optimized to bring the total number of cultures up to 12.
Again looking ahead to the cloning we saw that we would also need to increase the amount of pBR322 and pET28a vectors we had. For each of these we would need to create 3 8mL cultures to be able to perform an equimolar ligation. This all brought the total number of cultures we would need to create up to 63. The creating of the cultures was then performed in the following manner:
- All the following steps were performed in the vicinity of a blue flame to increase the sterility.
- 63 culture tubes were filled with 8mL of LB medium which had been prepared earlier.
- 8µL of 1000x Ampicillin antibiotics were then added to 60 of the culture tubes. 8µL of 1000x stock Kanamycin antibiotics were then added to the remaining three culture tubes. This was done as the pET28a vectors were resilient to Kanamycin and not to Ampicillin which all the other constructs were.
- The culture plates for each of the constructs were then retrieved. Instead of, as we did with the previous Protamine-1-Optimized constructs, pipetting a single culture into each of the culture tubes we now to decided to pick a culture and dissolve it in 15µL of water. From this 15µL solution 5µL could be pipetted into a culture tube. This meant that we would not have to pick 12 cultures from the plate per construct (or 9 for the Protamine-1-Optimized construct) but only 4 (or 3 for the Protamine-1-Optimized construct).
- For the pET28a culture, an eppendorf tube containing a glycerol stock of pET28a vectors in NB bacteria was retrieved and from the -80°C freezer and once thawed, 5µL was pipetted into each of the 3 culture tubes.
- For the pBR322 culture we would first have to transform the DNA into NB bacteria in the following fashion:
- Firstly the pBR322 vector DNA would have to be diluted as follows:
- Add 450µL of MilliQ water to the DNA to create a 100ng/µL stock solution.
- Take 1µL of the 100ng/µL stock solution and add it to a further 99µL of MilliQ water creating a 1ng/µL stock solution.
- Using this 1ng/µL stock solution the transformation could commense.
- 1µL of the stock solution was added to 20µL of NB bacteria.
- This solution was left on ice for 5 minutes before being placed in a 42°C water bath, heat shocking the bacteria.
- The bacterial solution was then placed on ice for a further 2 minutes before 80µL of SOC was added to the solution.
- Without placing on ice first the solution was then placed in a 37°C incubator for 60 minutes.
- After 60 minutes in the incubator the 20µL of the solution could be pipetted into each of the three pBR322 culture tubes.
- 60µL of SOC was subsequently added to the remainder of the bacterial solution so that it could be plated (on an Ampicillin agar plate) and in that way serve as a backup should the culture not grow.
- Firstly the pBR322 vector DNA would have to be diluted as follows:
- Now that all the culture tubes were complete they could be placed in a 37°C incubator overnight to grow.
Retaining the DNA
As there were 63 culture tubes it was decided to perform the miniprep protocol, used to retain the DNA from the cultures, on the cultures in two separate bulks. The first bulk would see the retention of DNA from the 1ETF, 1PJN1 and Poly(Aginine-Glycine) culture tubes. The second bulk would then deal with the Poly(Arginine-Serine), Protamine-1-Optimized, pBR322 and pET28a cultures. This did mean that the second bulk would have more time (several hours) to grow than the cultures of the first bulk. For both the bulks the same protocol was followed:
- Before beginning with the miniprep protocol 700µL of culture solution was added to 300µL of 50% Glycerol solution. This was done for one culture from each construct so that should something go wrong we would no longer have to worry about plating the desired constructs.
- To start the miniprep protocol the culture tubes were spun down in a centrifuge for 10 minutes at 3700rpm causing the bacterial cells with the DNA inside to form pellets isolating it from the growth medium.
- The supernatant that formed above the pellets was discarded and the pellets themselves were resuspended in 250µL of P1 buffer. The tubes were gently shaken by hand until the entire pellet had resuspended.
- The suspension was then transferred by means of pipetting into a smaller (1.5mL) eppendorf tube.
- 250µL of P2 buffer was then added to the eppendorf tube and the solution was inverted by hand until it had turned a clear blue.
- Within 5 minutes of adding the P2 buffer 350µL of N3 buffer was added to the solution and the eppendorf tube was inverted again until the solution was clear again.
- Once clear the eppendorf tube was placed in the centrifuge and spun for 10 minutes at 13000rpm seperating the bacterial cells from the DNA which remained suspended in the solution. The bacterial cells formed a pellet in the bottom of the eppendorf tube. (Unfortunately the pellet had not completely formed after 10 minutes so the eppendorf tube was spun for a further minute, again at 13000rpm.)
- As soon as the centrifuge had stopped and we could see that the pellet had properly formed, the supernatant was poured off into a special QIAcolumn. The pellet could be discarded in the bio-hazard waste.
- The QIAcolumn was subsequently centrifuged for one minute at 13000rpm, during which the DNA vector bound to the column meaning the flow through could be discarded.
- 750µL of PE buffer was then added to the QIAcolumn before centrifuging it for another minute at 13000rpm. The flow through was discarded.
- To ensure that all the PE buffer had passed through the column it was then spun yet again for one minute at a speed of 13000rpm. Yet again the flow through could be discarded.
- The column itself was then placed above a sealable 1.5mL eppendorf tube so that the DNA could be contained and stored. To obtain the DNA from the column 36µL of MilliQ water was pipetted onto the very centre membrane of the column after which it was spun down for one minute at 13000rpm. The flow through now sat in the 1.5mL eppendorf tube and contained the DNA vectors. (It would appear that for the 1ETf and 1PJN1 cultures 72µL of water was used instead of the desired 36µL meaning we had total volume for these tow samples which was twice as large as for the other cultures.)
Nanodrop Test
To determine how much DNA we had acquired for each of the constructs a nanodrop test could be performed. However before doing so all the DNA from a single construct was combined so that we had an indication of how much DNA we had in total. For the Protamine-1-Optimized sample this combination included the 3 50&mico;L solutions we had obtained previously. The results of this nanodrop test are given below:
- 432µL of Poly(Arginine-Glycine) with a concentration of 265.1ng/µL.
- 432µL of Poly(Arginine-Serine) with a concentration of 262.3ng/µL.
- 474µL of Protamine-1-Optimized with a concentration of 209.3ng/µL.
- 864µL of 1ETF with a concentration of 225.1ng/µL.
- 864µL of 1PJN1 with a concentration of 120.5ng/µL.
- 108µL of pBR322 with a concentration of 108.4ng/µL.
- 108µL of pET28a with a concentration of 62.1ng/µL.
Week 3
Digestion
Previously we had increased the amount of DNA we had to our disposal by culturing bacteria with containing our constructs and retaining the multiplied DNA using the miniprep method. Each of the five constructs we had designed and received (Protamine-1-Optimized, 1PJN1, 1ETF, Poly(Arginine-Serine) and Poly(Arginine-Glycine)) had successfully been multiplied along with the two vectors (pET28a and pBR322) in which we wished to insert our constructs. To insert our constructs into the two vectors we would first need to digest all the constructs and the vectors using the correct restriction enzymes and then ligate the insert and the vector. Today was focussed around the digestion.
Construct Digestion
As the our desired inserts for the ligation were in the pUC57-Simple vector we would need to perform digestion on all the vectors and then purify the product thereof to obtain the desired insert. However, due to the relatively large size of the inserts we could not do this using the standard PCR purification kit. This meant that purification would have to be done on basis of gel extraction. For this we would need a larger amount of DNA to start with to ensure a sufficient yield upon completion. It was decided that loading 10µg of DNA onto the gel for each construct would be adequate. Insert would be adequate. However for each construct two inserts would be created so each construct would have to provide 2 * 10µg of DNA for loading onto the gel.
For the digestion the following were added together in a PCR eppendorf tube:
| Construct | 5µg DNA (µL) | CutSmart Buffer (µL) | Amount of each Enzyme (µL) | MilliQ water (µL) |
|---|---|---|---|---|
| Protamine-1-Optimized | 23.9 | 5 | 0.7 | 20.4 |
| 1ETF | 22.2 | 5 | 0.7 | 22.1 |
| 1PJN1 | 41.5 | 5 | 0.7 | 2.8 |
| Poly(Arginine-Glycine) | 18.9 | 5 | 0.7 | 25.4 |
| Poly(Arginine-Serine) | 19.1 | 5 | 0.7 | 25.2 |
For each of the 5 constructs the solutions in the above table were prepared with the exception of the enzymes a total of four times. This provided us with 20µg of DNA in total per construct spread out equally across 4 seperate PCR tubes. To two of these the 0.7µg of the NheI and 0.7µg of the XhoI restriction enzymes were added. The product of the restriction with these enzymes would cut out just the protein sequence from the construct allowing us to ligate that into the pET28a vector for aerobic expression. To the remaining two eppendorf tubes for each of the constructs 0.7µg of the EcoRI and 0.7µg of the HindIII restriction enzymes were added. This restriction would cut our entire construct from the pUC57-simple vector allowing us to ligate that into the pBR322 vector and test our construct anaerobically.
After adding the enzymes the solutions were quickly mixed and placed in the PCR apparatus.
Vector Digestion
Rather more simple than the construct digestion the vectors too needed to be digested. As we would only require 500µg of DNA as a result of this digestion it was not necessary to use 5µg of DNA so it was decided only 2µg would be used per vector. The following solutions were made:
| Vector | 2µg DNA (µL) | CutSmart Buffer (µL) | Amount of each Enzyme (µL) | MilliQ water (µL) |
|---|---|---|---|---|
| pBR322 | 32.2 | 5 | 0.7 | 12.1 |
| pET28a | 18.5 | 5 | 0.7 | 25.8 |
0.7µL of the NheI and 0.7µL of the XhoI restriction enzymes were added to the pET28a vector solution and 0.7µL of the EcoRI and 0.7µL of the HindIII restriction enzymes were added to the pBR322 vector solution. These solutions were then placed in the same PCR apparatus as the construct solutions and the following PCR protocol was run for both the constructs and the vectors simultaneously:
- Reaction Step: 37°C for 60 minutes
- Enzyme Inactivation Step: 80°C for 20 minutes
- Storage Step: 4°C
PCR Purification for the Constructs
Due to the large size of the insert produced by the digestion of the constructs a gel extraction would be used to select the appropriate sections of the digestion product of the constructs. To isolate the correct segments on the gel the following steps were taken:
- A 1% electrophoresis gel was created with 10 large lanes (each lane large enough for a 120µL sample).
- For each of the constructs the two PCR solutions containing the same enzymes were pipetted into a single tube creating a single 100µL solution.
- To each tube containing a 100µL solution 20µL of 6x stock solution loading dye was added bringing the total volume up to 120µL.
- Each of these 120µL samples were then brought over into one of the 10 available lanes.
- The gel was the "run" for 60 minutes with a voltage of 100V.
- After "running" the gel it was removed from its container and placed on a UV light. Here we were able to discern between the different bands. For each of the digestion results the smaller DNA segment was desired so the lowest band in each lane of the gel was cut from the gel and placed in an eppendorf tube.
- The 10 eppendorf tubes containing the gel cut-outs were then placed in the -20°C freezer for storage until the real DNA extraction would take place.
PCR Puricfication for the Vectors
For the vectors a simple QIAquick PCR purification kit could be used to purify the digestion product as the cut out was smaller than 100 bases for each of the two vectors. The purification was carried out according to the following protocol:
- To the PCR product 5 volumes of PB buffer were added (In our case this was 250µL of PB buffer as we had 50µL of PRC product.)
- This solution was placed in a QIAquick spin column and spun for 60 seconds at 13000 rpm. The flow through could be discarded.
- Now 750µL of PE buffer could be added to the column before spinning it down once more for 60 seconds at 13000 rpm.
- Having discarded the flow through the column was spun for a further 60 seconds again at 13000 rpm to remove the last of the PE buffer.
- The column was then placed into a sealable eppendorf tube before adding 42µL of MilliQ water to the very centre membrane of the spin column and leaving it to stand for one minute.
- The column was now placed in the centrifuge and spun for 60 seconds at 13000 rpm producing a solution containing the desired DNA.
Nanodrop Results
To test how successful the entire process of restriction had been for the vectors the purification products were analysed using the nanodrop spectrometer. the results are given below:
- pBR322 had a concentration of 11.6ng/µL.
- pET28a had a concentration of 7.9ng/µL.
Having eluded both of these samples in just 42µL of water meant that we did not have enough vector DNA for the ligation later on (we were aiming for 500ng). To solve this the entire restriction was performed again for the vectors. This meant remaking the solutions for the digestion and restarting the PCR. As it was late in the day, the solutions were prepared, the enzymes were added and the PCR was started. The PCR machine was set to return to 4°C to store the restriction product overnight and the PCR purification could then be performed the next day along with another nanodrop analysis, hopefully with better results.
Continuing with the Purification
On August 5th digestion was performed on five of our ordered constructs, and on the two vectors in which we would like to place our constructs. At the end of the day the digestion products of the constructs were still within the gel cut outs which had been stored in a -20°C freezer. The constructs had been purified but the yield had been too low for ligation so the digestion was performed once more. Today then, before moving on to the ligation, the constructs would have to be extracted from the gel and the vectors would also have to be purified.
Vector Purification
The purification of the vectors was performed in the same manner this time round as it was the previous time, following the protocol of the QIAquick PCR purification kit. To prevent falling into repetition this protocol will not be discussed here but can be found in the journal entry for August 5th. The results of this purification however, gotten through a nanodrop measurement are given below:
- pBR322 had a concentration of 15.3ng/µL.
- pET28a had a concentration of 10.7ng/µL.
Needing these digested vectors for the ligation later on, we had hoped for a yield of at least 500ng of DNA per vector. Based on these results and an estimated volume of around 38µL this meant that we had achieved our goal for the pBR322 vector. For the pET28 factor however this was not the case. Fortunately we had not discarded the solutions from the previous pET28a digestion meaning that their combined volumes and concentrations would be sufficient for the ligation. From now on pET28a(1) will refer to the pET28a sample digested on August 5th with a concentration of 9.8ng/µL and pET28a(2) will refer to the pet28a sample digested today with a concentration of 10.7ng/µL.
Construct Purification
Our constructs had been run on gels to separate the different products formed during the digestion. The desired bands had been cut out from these gels and had been stored in the -20°C freezer overnight. Now we were ready to perform the gel extraction, retrieving the DNA from these gel cut outs. This was done following the QIAquick gel extraction protocol which is explained here:
- First all the samples were weighed. A single QIAcolumn can be used for up to 400ng of gel, as all our cut-outs were almost double this they were disected into two almost equal parts and spread over two separate eppendorf tubes. The weight of the gel in each tube was noted.
- To each tube 3 volumes of QG buffer was added. A volume can be defined as the weight of the gel in mg transferred into µL. SO a gel weighing 400mg would have a volume of 400µL, so 1200µL of QG would be added.
- The eppendorf tubes containing the gel cut-outs and the QG buffer were placed in an incubator set at 50°C for 10 minutes, whilst vortexing the samples at regular (every 2 minutes) intervals until the gel had completely dissolved.
- To these tubes 1 volume of isopropanol was added. As the addition of this isopropanol would not fit in a single eppendorf tube the solution in each of the tubes was split equally across two separate eppendorf tubes. To each of these separate eppendorf tubes half a volume of isopropanol was added.
- One of the two tubes was then brought over into a QIAquick column and placed in the centrifuge where it was spun down for 60 seconds at 13000 rpm. After discarding the flow through the second of the two tubes was then brought over into the QIAquick column and spun down in the same fashion. Once more the flow through was discarded.
- Now 750µL of PE buffer was added to the column before centrifuging at 13000 rpm for 60 seconds. After discarding the flow through the same amount was added and once more and again centrifuged.
- Removing the flow through the column was then centrifuged again for 60 seconds at 13000 rpm removing the last of the PE buffer.
- After bringing the column across into a sealable eppendorf tube 36µL of MilliQ water was pipetted onto the very centre membrane of the column which was then let to stand for 60 seconds. Subsequently the column was spun down at 13000 rpm for 60 seconds. The DNA is now in the flow through.
- Remembering that we split the gel in half in the very first stage of this protocol, the two solutions containing the same DNA were combined so that a total volume of maximally 72µL of each insert was formed.
Nanodrop Results
To check the concentration of the inserts we had acquired a nanodrop test was performed using the spectrometer. The results are given below. Remember that for each construct, two different digestions were performed, one for ligation into pET28a and one for ligation into pBR322.
| Construct | pET28a Digest (ng/µL) | pBR322 Digest (ng/µL) |
|---|---|---|
| Protamine | 2.6 | 9.6 |
| 1PJN1 | 5.0 | 10.5 |
| 1ETF | 7.0 | 11.1 |
| Poly(Arginine-Glycine) | 11.8 | 16.0 |
| Poly(Arginine-Serine) | 8.3 | 13.4 |
Ligation
Each of our five constructs (Protamine-1-Optimized, 1PJN1, 1ETF, Poly(Arginine-Glycine) and Poly(Arginine-Serine)) restriction was performed in two ways. The first cutting at the NheI and XhoI restriction sites allowing us to place just the protein sequence into the pET28a vector. The second restriction cut at the EcoRI and HindIII sites, thus allowing us to ligate the entire construct (protein sequence along with promoters, terminators and His-tags) into the pBR322 vector.
Before being able to prepare the ligation solution it was necessary to perform a number of calculations. For each vector we added to the solution we wished to add five times as many inserts. This would ensure that our inserts would be ligated into the vectors. To start with we calculated how many µL of our digested vectors would need to be added to the ligation solution to obtain 100ng of vector DNA.
| Vector | Concentration (ng/µL) | 100g DNA (µL) | Number of Bases in Vector |
|---|---|---|---|
| pBR322 | 15.3 | 6.6 | 4330 |
| pET28a(1) | 9.8 | 10.2 | 5296 |
| pET28a(2) | 10.7 | 9.4 | 5296 |
By dividing 100ng by the number of bases in a single vector and multiplying that number by the number of bases in a single insert it can be determined how many ng of the insert is needed for molar equality (having an equal amount of vectors as there are inserts).
By then dividing the amount of DNA needed for molar equality (in ng) by the concentration of each insert the volume (in µL) of insert solution needed would be given. As we did not want molar equality but five times as many inserts as there are vectors this volume could be multiplied by five.
The calculations for the pBR322 inserts are given here:
| Construct | Number of Bases | Concentration (ng/µL) | ng Needed for Molar Equality (ng) | Volume Needed for Molar Equality (µL) | Volume Needed for 5x Molar Equality (µL) |
|---|---|---|---|---|---|
| Protamine-1-Optimized | 489 | 9.2 | 11.3 | 1.23 | 6.2 |
| 1PJN1 | 726 | 10.5 | 16.8 | 1.6 | 8.0 |
| 1ETF | 756 | 11.1 | 17.5 | 1.58 | 7.9 |
| Poly(Arginine-Glycine) | 771 | 16.0 | 17.8 | 1.11 | 5.6 |
| Poly(Arginine-Serine) | 771 | 13.4 | 17.8 | 1.32 | 6.6 |
For the pET28a vector neither of the two digested samples had enough DNA to provide 100ng for ligation with each of the five constructs. Therefore it was decided to use the pET28a(1) digestion product for the Poly(Arginine-Glycine) and Poly(Arginine-Serine) inserts. The pET28a(2) digestion product would then be used for the remaining three ligations (with Protmaine-1-Optimized, 1PJN1 and 1ETF).
The calculations are given here for the pET28a(1) ligation:
| Construct | Number of Bases | Concentration (ng/µL) | ng Needed for Molar Equality (ng) | Volume Needed for Molar Equality (µL) | Volume Needed for 5x Molar Equality (µL) |
|---|---|---|---|---|---|
| Poly(Arginine-Glycine) | 447 | 11.8 | 8.44 | 0.72 | 3.6 |
| Poly(Arginine-Serine) | 447 | 8.3 | 8.44 | 1.02 | 5.1 |
And for the pET28a(2) ligation here:
| Construct | Number of Bases | Concentration (ng/µL) | ng Needed for Molar Equality (ng) | Volume Needed for Molar Equality (µL) | Volume Needed for 5x Molar Equality (µL) |
|---|---|---|---|---|---|
| Protamine-1-Optimized | 165 | 2.6 | 3.11 | 1.2 | 6.0 |
| 1PJN1 | 402 | 5.0 | 7.6 | 1.52 | 7.6 |
| 1ETF | 432 | 7.0 | 8.2 | 1.2 | 5.9 |
Having determined the volumes of insert and vector we would need for the ligation reactions, the ligation solutions could be prepared. In addition to the vectors and inserts we would also need to add a buffer, MilliQ water and Ligase. All reaction solutions would end up equal to 20µL. The table below shows the amounts used to prepare the pBR322 vector ligation samples:
| Construct | Amount of Vector pBR322 (µL) | Amount of Insert (µL) | 10x Buffer (µL) | MilliQ Water (µL) | Ligase (µL) |
|---|---|---|---|---|---|
| Protamine-1-Optimized | 6.6 | 6.2 | 2 | 4.2 | 1 |
| 1PJN1 | 6.6 | 8.0 | 2 | 2.4 | 1 |
| 1ETF | 6.6 | 7.9 | 2 | 2.5 | 1 |
| Poly(Arginine-Glycine) | 6.6 | 5.6 | 2 | 4.8 | 1 |
| Poly(Arginine-Serine) | 6.6 | 6.6 | 2 | 3.8 | 1 |
Below are give the solution quantities for the pET28a(1) and pET28a(2) vector ligations respectivly:
| Construct | Amount of Vector pET28a(1) (µL) | Amount of Insert (µL) | 10x Buffer (µL) | MilliQ Water (µL) | Ligase (µL) |
|---|---|---|---|---|---|
| Poly(Arginine-Glycine) | 10.2 | 3.6 | 2 | 3.1 | 1 |
| Poly(Arginine-Serine) | 10.2 | 5.1 | 2 | 1.7 | 1 |
| Construct | Amount of Vector pET28a(2) (µL) | Amount of Insert (µL) | 10x Buffer (µL) | MilliQ Water (µL) | Ligase (µL) |
|---|---|---|---|---|---|
| Protamine-1-Optimized | 9.4 | 6.0 | 2 | 1.6 | 1 |
| 1PJN1 | 9.4 | 7.6 | 2 | 0.0 | 1 |
| 1ETF | 9.4 | 5.9 | 2 | 1.7 | 1 |
Having prepared the above solutions, and having added the Ligase only at the very last moment, the solutions were placed in the PCR apparatus ready for the ligation. The ligation protocol was set as follows:
- 37°C for 120 minutes for the ligation reaction.
- 65°C for 10 minutes for the Ligase inactivation.
- 4°C FOREVER to store the ligation products until they would be used again.
The samples were left in the PCR apparatus overnight.
Preparing Agar Plates
Before leaving, and in anticipation of the coming steps we decided to prepare a number of Ampicillin agar plates. The agar solution itself was made by mixing the following:
- 5g Peptones
- 5g NaCl
- 2.5g Yeast Extract
- 7.5g Agar
- 500mL Demineralised Water
By placing the above solution in the autoclave the agar was produced. Once cool 500µL of Ampicillin was added to the solution whilst being stirred. In the vicinity of a blue bundsen flame 25 Ampicilling agar plates were poured which were then left to harden before being placed in the 4°C refrigerator.
Transformation
Having left the ligation products in the PCR apparatus at 4°C overnight we were now able to transform these into NoveBlue bacteria ready to plate them. This plating will allow us to perform a ColonyPCR later on to check the efficiency of our ligation. It would also allow us to replicate, duplicate and sequence correctly ligated vectors and inserts. For the transformation we transformed all 10 ligation products (each of the five constructs in both the vectors) using the same transformation protocol:
- All steps were performed in the vicinity of a blue flame to increase sterility.
- 1µL of each ligation product was pipetted into 20µL of NovaBlue bacteria.
- These were left to stand on ice for 5 minutes.
- Then to heat shock the samples they were placed in a water bath at 42°C for 30 seconds.
- The samples were subsequently returned to ice for 2 minutes.
- Then 80µL of SOC solution was added to each of the samples. After this they would not be allowed to return to ice.
- The samples were then placed in an incubator set to 37°C for 60 minutes.
Apart from transforming the ligation products three controls were also prepared. This was done to check that the restriction of the vectors had been successful. To make the control samples made samples of 20µL consisting of the same amount of vector as used in the ligation solutions. The remaining volume (up to 20µL) was then filled with water. The transformation protocol of these controls follows that which was used for the ligation products.
Plating
Once the transformations had been incubated for an hour they were ready for plating to allow colonies to grow on the plates. The samples which had been ligated into the pET28a vectors and the two pET28a vector controls were plated onto agar platescontianing Kanamycin antibiotics. The samples which had been ligated into pBR322 vectors and the pBR322 vector control were plated onto agar plates containing Ampicillin antibiotics. For the plating the following protocol was used:
- In the vicinity of a blue bunsen flame the entire sample (101µL) was pipetted onto the centre of the agar plate.
- A spreader which had been stored in 100% Ethanol was lit in the flame burning off the Ethanol and sterilizing the spreader.
- The sterile spreader was allowed to cool before being placed onto the agar plate and spreading out the bacterial sample equally.
The plates had now been spread with the transformations of the ligation products and were placed in the 37°C incubator overnight. Here they could grow and with luck, be used for ColonyPCR the next day.
Colony Picking and PCR
Having plated NovaBlue bacteria containing our own ligated vectors (Each of our five constructs: Protamine-1-Optimized, 1PJN1, 1ETF, Poly(Arginine-Glycine) and Poly(Arginine-Serine), were ligated into both pBR322 and pET28a vectors) we could now pick a number of these samples, check the ligation products using PCR and culture those ligation products which had been successful. Due to the late delivery of the pBR322 primers it was decided to perform the colony PCR on just the pET28a ligations. The pBR322 agar plates would be stored at 4°C until the primers arrived. The following protocol was then used to perform the colony picking and PCR:
- All steps up to the PCR itself were performed in the vicinity of a blue bunsen flame for increased sterility.
- From each agar plate (each plate containing one of the constructs ligated with the pET28a vector) 5 seperate colonies were picked and dissolved in 15µL of sterile water. We had also plated the two separately digested pET28a factors as controls and from these plates a single colony was also picked and dissolved in 15µL of sterile water. Photos of these plates are visible below:
- After dissolving the pickings they were ready for PCR. Before starting the PCR protocol the following solutions had to be made:
- 9µL of sterile water.
- 1.25µL of 10µM T7 Fwd Primer.
- 1.25µL of 10µM T7 Rw Primer.
- 12.5µL of Kappa2G 2x solution.
- 1µL of the picking solution.
- Having mixed the above, adding the Kappa2G last, the PCR tubes were placed in the PCR and run with the following protocol:
- Initial Denaturation: 95°C for 3 minutes.
- The following steps should be performed in this order, repeating the process a total of 34 times.
- Denaturation: 95°C for 15 seconds.
- Annealing: 58°C for 15 seconds.
- Extension: 72°C for 20 seconds.
- Final Extension: 72°C for 10 minutes.
- Storage: 4°C FOREVER.
- Having completed the PCR protocol 4µL of 6x Loading Dye was added to each of the PCR solutions. Subsequently 18µL of each was loaded onto a 1.5% agarose electrophoresis gel which was prepared by mixing the following:
- 1.8g of Agarose.
- 120mL of 1x TAE buffer
- 12µL of CyberSafe.
- The gel was then run at 100V for 75 minutes before placing the gel into a UV spectrometer. Here we could analyse the gel and decide which of the ligation products were successful. Of each of the five constructs two successfully ligated bacteria colonies were selected and these would then be grown in a larger culture. The gel result is shown below.
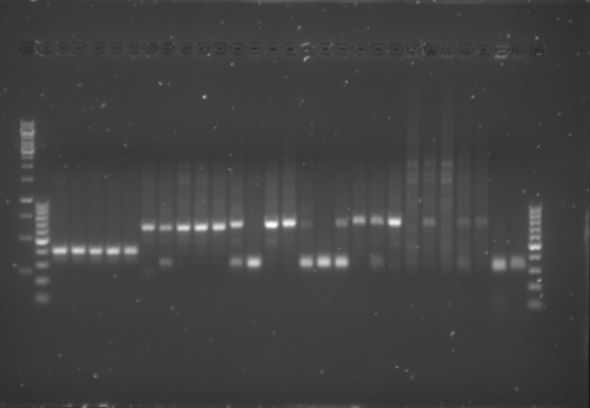
Culturing the Ligation Products
Having analyzed the gel and having chosen two successful ligations for each of the constructs these were then cultured. This was done in the following manner:
- All below steps were performed near a blue bunsen flame to increase sterility.
- Culture tubes were filled with 8mL of LB medium.
- To each of the culture tubes 8µL of Kanamycin was added.
- 5µL of the picked colony solution was pipetted into the culture medium.
- The culture tubes were then placed in the 37°C incubator overnight.
Miniprep
On August 8th a number of constructs were chosen to be cultured, multiplying the number of correctly ligated DNA vectors we had available to ourselves. To retain this DNA the miniprep protocol would be performed on each of the cultures:
- To start the miniprep protocol the culture tubes were spun down in a centrifuge for 10 minutes at 3700rpm causing the bacterial cells with the DNA inside to form pellets isolating it from the growth medium.
- The supernatant that formed above the pellets was discarded and the pellets themselves were resuspended in 250µL of P1 buffer. The tubes were gently shaken by hand until the entire pellet had resuspended.
- The suspension was then transferred by means of pipetting into a smaller (1.5mL) eppendorf tube.
- 250µL of P2 buffer was then added to the eppendorf tube and the solution was inverted by hand until it had turned a clear blue.
- Within 5 minutes of adding the P2 buffer 350µL of N3 buffer was added to the solution and the eppendorf tube was inverted again until the solution was clear again.
- Once clear the eppendorf tube was placed in the centrifuge and spun for 10 minutes at 13000rpm seperating the bacterial cells from the DNA which remained suspended in the solution. The bacterial cells formed a pellet in the bottom of the eppendorf tube. (Unfortunately the pellet had not completely formed after 10 minutes so the eppendorf tube was spun for a further minute, again at 13000rpm.)
- As soon as the centrifuge had stopped and we could see that the pellet had properly formed, the supernatant was poured off into a special QIAcolumn. The pellet could be discarded in the bio-hazard waste.
- The QIAcolumn was subsequently centrifuged for one minute at 13000rpm, during which the DNA vector bound to the column meaning the flow through could be discarded.
- 750µL of PE buffer was then added to the QIAcolumn before centrifuging it for another minute at 13000rpm. The flow through was discarded.
- To ensure that all the PE buffer had passed through the column it was then spun yet again for one minute at a speed of 13000rpm. Yet again the flow through could be discarded.
- The column itself was then placed above a sealable 1.5mL eppendorf tube so that the DNA could be contained and stored. To obtain the DNA from the column 42µL of MilliQ water was pipetted onto the very centre membrane of the column after which it was spun down for one minute at 13000 rpm. The flow through now sat in the 1.5mL eppendorf tube and contained the DNA vectors.
Nanodrop Results
A useful step now, having acquired our ligated DNA vectors would be to send them off for sequencing to confirm that we had indeed ligated and cultured the correct vectors and inserts. Before being able to do this, we would need to determine the concentrations of each of the miniprep products.
| Vector | Colony Picking Number | Concentration (ng/µL) |
|---|---|---|
| Protamine-1-Optimized | 1 | 60.0 |
| Protamine-1-Optimized | 2 | 65.9 |
| 1PJN1 | 3 | 72.1 |
| 1PJN1 | 4 | 66.6 |
| 1ETF | 3 | 64.1 |
| 1ETF | 4 | 72.8 |
| Poly(Arginine-Glycine) | 3 | 73.5 |
| Poly(Arginine-Glycine) | 5 | 117.8 |
| Poly(Arginine-Serine) | 2 | 206.7 |
| Poly(Arginine-Serine) | 5 | 57.8 |
Vector Sequencing
To sequence each of the vectors we sent off samples of our DNA to an external sequencing company. For this company to fully sequence each of our vectors we had to create two solutions for each vector we wished to sequence, one with the T7 FWD primer and one with the T7 RW primer, both with a concentration of 10µM. The second requirement was that we added between 400ng and 600ng of our DNA vectors. Unfortunately the maximal volume of the entire solution was 7µL, so for constructs with a concentration lower than 67ng/µL the minimal amount of DNA desired for sequencing was not reached. This led to the creation of the following solutions, with the same solution being made for both primers:
| Vector | Colony Picking Number | Volume Vector Solution (µL) | Water (µL) | Primer (µL) |
|---|---|---|---|---|
| Protamine-1-Optimized | 1 | 6.0 | 0.0 | 1.0 |
| Protamine-1-Optimized | 2 | 6.0 | 0.0 | 1.0 |
| 1PJN1 | 3 | 6.0 | 0.0 | 1.0 |
| 1PJN1 | 4 | 6.0 | 0.0 | 1.0 |
| 1ETF | 3 | 6.0 | 0.0 | 1.0 |
| 1ETF | 4 | 6.0 | 0.0 | 1.0 |
| Poly(Arginine-Glycine) | 2 | 6.0 | 0.0 | 1.0 |
| Poly(Arginine-Glycine) | 5 | 4.0 | 2.0 | 1.0 |
| Poly(Arginine-Serine) | 3 | 3.0 | 3.0 | 1.0 |
| Poly(Arginine-Serine) | 5 | 6.0 | 0.0 | 1.0 |
Having prepared these solutions in small PCR eppendorf tubes they were packaged and sent off for sequencing.
Colony Picking and PCR
On August 8th we had performed the colony picking and PCR for the constructs which had been ligated into the pET28a vector. As we did not have the primers for the pBR322 vector those colony plates were stored at 4°C. Today however the pBR322 primers had arrived and we could perform the colony pickings for the ligated pBR322 vectors. It should be noted that the primers we used for the pBR322 vector had been designed ourselves with the following sequences:
| pBR322 FWD Primer | 5’- CCC GAA AAG TGC CAC CTG -3’ |
| pBR322 RW Primer | 5’- GT GCC GAG GAT GAC GATG -3’ |
To then perform the colony PCR the following solutions had to be created:
- Much like for the pET28a ligated vectors we picked five colonies for each of the five constructs ligated into the pBR322 vector. These pickings were dissolved in 5µL of sterile water. Along with the five pickings per construct we also picked one colony from the control plate, which contained just the pBR322 vector. Photos of the agar plates are shown below:

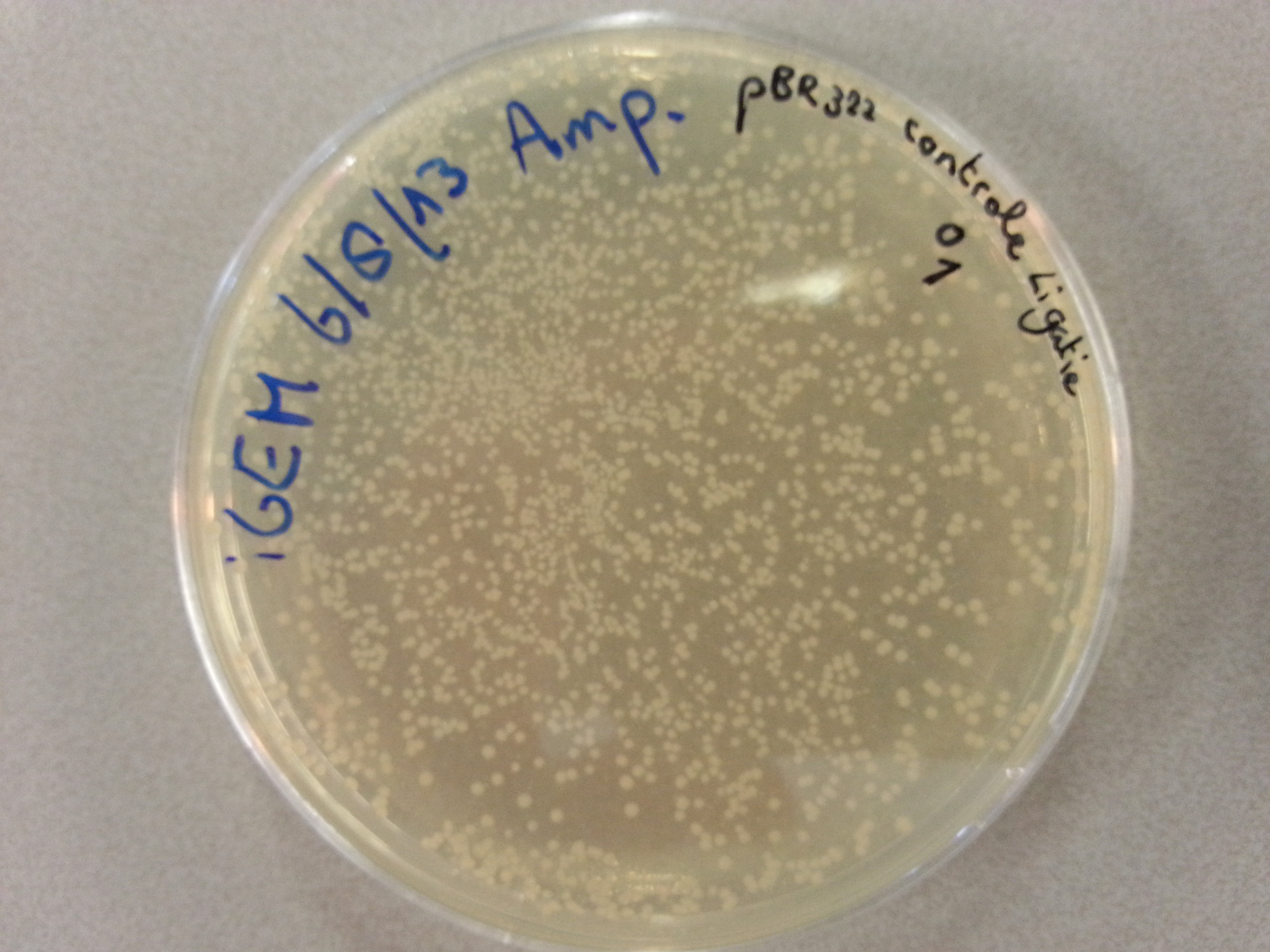
- We could now go ahead and prepare our PCR solutions by mixing the following:
- 9µL of sterile water.
- 1.25µL of the FWD primer.
- 1.25µL of the RW Primer.
- 1µL of the pBR322 ligated vector colony picking solution.
- 12.5µL of Kapa2G.
- We made sure to only add the Kapa2G at the very end just before placing the solutions in the PCR.
Having made the above solutions they were brought across to the PCR apparatus which then performed the following PCR protocol:
- Initial Denaturation: 95°C for 3 minutes.
- The following steps should be performed in this order, repeating the process a total of 34 times.
- Denaturation: 95°C for 15 seconds.
- Annealing: 58°C for 15 seconds.
- Extension: 72°C for 20 seconds.
- Final Extension: 72°C for 10 minutes.
- Storage: 4°C FOREVER.
Having completed the PCR protocol 4µL of 6x Loading Dye was added to each of the PCR solutions. Subsequently 18µL of each was loaded onto a 1.5% agarose electrophoresis gel which was prepared by mixing the following:
- 1.8g of Agarose.
- 120mL of 1x TAE buffer
- 12µL of CyberSafe.
The gel was then run at 100V for 75 minutes before placing the gel into a UV spectrometer. Here we could analyse the gel and decide which of the ligation products were successful. As it was a Friday afternoon there was no possibility to culture the correctly ligated vectors so these were stored in the 4°C refrigerator and left there until they could be cultured.
Week 4
Transforming and Plating New Constructs
Today we began work on four new constructs which had arrived late last week. The constructs that we had received were: EGFP, 1G70, Poly(Threonine-Lysine) and Poly(Lysine-Serine). For more information of these constructs please see the wetlab preparation page. The first step to be performed on each of the constructs was the preparing of two dilations. The first a 200ng/µL dilation made by adding 20µL of MilliQ water to the standard 4µg of DNA we recieved for each construct. From this dilution we then took 1µL and added it to another 199µL of MilliQ water forming a 1ng/µL solution which we could use for the transformation:
- All steps for the transformation were performed near a blue flame for sterility.
- Start by adding 1µL of the 1ng/µL solution to 20µL of NB bacteria.
- Place the bacteria on ice and let them stand for 5 minutes.
- Heat shock the bacteria by placing them in a water bath of 42°C for 30 seconds before returning them to ice for two minutes.
- Now add 80µL of SOC to each sample and place the samples in a 37°C incubator for an hour. The samples should not (and were not) returned to ice.
This concludes the transformation stage of the constructs which are now enclosed within the NB bacteria. After incubation the bacteria can therefore be plated onto Ampicillin agar plates.
- In the vicinity of a blue bunsen flame place pipet the entire solution of bacterial cells and SOC onto the centre of the plate.
- Using a sterile spreader spread out the solution over the gel surface.
- The plates can now be incubated at 37°C for an overnight growth.
This concludes the work on the new constructs for today.
Transforming and Plating Old Constructs
Last week a number of constructs were digested and ligated into pET28a. These new vectors were then multiplied and retained by performing a culture and miniprep respectively. These new vectors could now be transformed into BL21 bacteria so that expression could be induced using IPTG. Before embarking on the transformation however the following solutions had to be made. As you can see we no longer use all 10 ligated constructs to reduce the work load and will continue with one of each. The concentrations used were determined last week. These solutions will create 1ng/µL solutions of each of the ligated vector solutions.
| Vector | Colony Picking Number | Concentration (ng/µL) | Sterile Water (µL) | Vector Solution (µL) |
|---|---|---|---|---|
| Protamine-1-Optimized | 1 | 60.0 | 59.0 | 1.0 |
| 1PJN1 | 3 | 72.1 | 71.1 | 1.0 |
| 1ETF | 4 | 72.8 | 71.8 | 1.0 |
| Poly(Arginine-Glycine) | 3 | 73.5 | 72.5 | 1.0 |
| Poly(Arginine-Serine) | 5 | 57.8 | 56.8 | 1.0 |
Having prepared the above solutions the transformation could be started:
- All steps for the transformation were performed near a blue flame for sterility.
- Start by adding 1µL of the 1ng/µL solution to 20µL of BL21 bacteria.
- Place the bacteria on ice and let them stand for 5 minutes.
- Heat shock the bacteria by placing them in a water bath of 42°C for 30 seconds before returning them to ice for two minutes.
- Now add 80µL of SOC to each sample and place the samples in a 37°C incubator for an hour. The samples should not (and were not) returned to ice.
This concludes the transformation stage of the constructs which are now enclosed within the BL21 bacteria. After incubation the bacteria can therefore be plated onto Kanamycin agar plates.
- In the vicinity of a blue bunsen flame place pipet the entire solution of bacterial cells and SOC onto the centre of the plate.
- Using a sterile spreader spread out the solution over the gel surface.
- The plates can now be incubated at 37°C for an overnight growth.
This concludes the work on the ligated pET28a constructs for today.
Creating Small Cultures
As we were unable to produce small cultures of the old constructs ligated into the pBR322 vector last week this too would be performed today. Before beginning a new batch of LB medium was prepared:
- The LB medium was created by mixing the following:
- 10g Peptones
- 10g NaCl
- 5g Yeast Extract
- 1L Demineralised Water
- After adding together the above, the solution was autoclaved and left to cool.
One the LB medium was ready for use the culture preparation could be performed:
- Using the electrophoresis gel from the previous week two ligated vectors were chosen to cultured for each of the five constructs.
- This meant that 10 culture tubes would have to be filled with 8mL of LB medium each.
- To each tube a further 8µL of Ampicillin was added.
- Finally 8µL of the chosen construct was added and the culture tubes were placed in the 37°C incubator for overnight growth.
This concluded not only the work on the pBR322 ligated constructs for today, but also the rest of the days work.
Miniprepping ligated pBR322 vector cultures
On August 12th the pBR322 ligated constructs were placed in an incubator for overnight culture growth. Today we removed these cultured samples from the incubator, ready to retain the DNA. To do this the miniprep protocol would be used for each of the culture tubes. As a reminder, we had 10 culture tubes, 2 for each of the ligation constructs: Protamine-1-Optimized, 1PJN1, 1ETF, Poly(Arginine-Glycine), Poly(Arginine-Serine) ligated with pBR322. The miniprep went as follows:
- First the culture tubes were spun down in a centrifuge for 10 minutes at 3700G causing the bacterial cells with the DNA inside to form pellets isolating it from the growth medium.
- The supernatant that had formed above the pellets was discarded and the pellets themselves were resuspended in 250µL of P1 buffer. The tubes were gently shaken by hand until the entire pellet had resuspended.
- The suspension was then transferred by means of pipetting into a smaller (1.5mL) eppendorf tube.
- 250µL of P2 buffer was then added to the eppendorf tube and the solution was inverted by hand until it had turned a clear blue.
- Within 5 minutes of adding the P2 buffer 350µL of N3 buffer was added to the solution and the eppendorf tube was inverted again until the solution was clear again.
- Once clear the eppendorf tube was placed in the centrifuge and spun for 10 minutes at 13000rpm seperating the bacterial cells from the DNA which remained suspended in the solution. The bacterial cells formed a pellet in the bottom of the eppendorf tube. (Unfortunately the pellet had not completely formed after 10 minutes so the eppendorf tube was spun for a further minute, again at 13000rpm.)
- As soon as the centrifuge had stopped and we could see that the pellet had properly formed, the supernatant was poured off into a special QIAcolumn. The pellet could be discarded in the bio-hazard waste.
- The QIAcolumn was subsequently centrifuged for one minute at 13000rpm, during which the DNA vector bound to the column meaning the flow through could be discarded.
- 750µL of PE buffer was then added to the QIAcolumn before centrifuging it for another minute at 13000rpm. The flow through was discarded.
- To ensure that all the PE buffer had passed through the column it was then spun yet again for one minute at a speed of 13000rpm. Yet again the flow through could be discarded.
- The column itself was then placed above a sealable 1.5mL eppendorf tube so that the DNA could be contained and stored. To obtain the DNA from the column 42µL of MilliQ water was pipetted onto the very center membrane of the column after which it was spun down for one minute at 13000rpm. The flow through now sat in the 1.5mL eppendorf tube and contained the DNA vectors.
The following concentrations were then measured for each of the DNA samples:
| Vector | Colony Picking Number | Concentration (ng/µL) |
|---|---|---|
| Protamine-1-Optimized | 1 | 93.9 |
| Protamine-1-Optimized | 2 | 144.8 |
| 1PJN1 | 1 | 90.5 |
| 1PJN1 | 2 | 122.4 |
| 1ETF | 3 | 86.4 |
| 1ETF | 4 | 66.9 |
| Poly(Arginine-Glycine) | 1 | 71.4 |
| Poly(Arginine-Glycine) | 2 | 71.6 |
| Poly(Arginine-Serine) | 1 | 61.2 |
| Poly(Arginine-Serine) | 5 | 75.8 |
We later saw that the elution for the Protamin-1-Optimized, picking 2, solution had not been successful as the volume of retained DNA was a mere 10µL. This meant that from now on only one of the Protamine samples would be available.
Sequencing
Having retained the DNA vectors we could send off samples for sequencing. Like the previous time, for each of the samples 2 sequencing solutions would have to be prepared. One would contain 1µL of our self-designed pBR322 FWD primer and the other would contain1µL of our self-designed pBR322 RW primer. The following solutions were therefore made:
| Vector | Colony Picking Number | Volume Vector Solution (µL) | Water (µL) | Primer (µL) |
|---|---|---|---|---|
| Protamine-1-Optimized | 1 | 6.0 | 0.0 | 1.0 |
| Protamine-1-Optimized | 2 | 4.0 | 2.0 | 1.0 |
| 1PJN1 | 1 | 6.0 | 0.0 | 1.0 |
| 1PJN1 | 2 | 5.0 | 1.0 | 1.0 |
| 1ETF | 3 | 6.0 | 0.0 | 1.0 |
| 1ETF | 4 | 6.0 | 0.0 | 1.0 |
| Poly(Arginine-Glycine) | 1 | 6.0 | 0.0 | 1.0 |
| Poly(Arginine-Glycine) | 2 | 6.0 | 0.0 | 1.0 |
| Poly(Arginine-Serine) | 1 | 6.0 | 0.0 | 1.0 |
| Poly(Arginine-Serine) | 5 | 6.0 | 0.0 | 1.0 |
As mentioned there was only a small elution volume for the Protamine-1-Optimized, picking 2, DNA solution, so we could only prepare the FWD sequencing sample. For the reverse solution the DNA quantity was too low.
As we would only be needing the pBR322 constructs for anaerobic expression, which would only come after we had tested the CEST contrast of each construct, these DNA vectors were placed in the -20°C freezer and were to be left there until needed again in a later stage of this project.
Culturing New Constructs
On August 12th work began on four new constructs which had arrived: EGFP, 1G70, Poly(Threonine-Lysine) and Poly(Lysine-Serine). We had transformed each of the constructs into NovaBlue bacteria and had left these to grow overnight on Ampicillin agar plates. Today we would pick a number of grown colonies and culture them in small cultures. To do this the following steps were taken:
- All the following steps were performed near a blue flame for increased sterility.
- Fill 16 cultures tubes with 8mL of LB medium.
- Add 8µL of Ampicillin to each of the culture tubes.
- From each of the four culture plates 2 colonies were picked and dissolved in 15µL of sterile water. Pictures of the plates are given below.
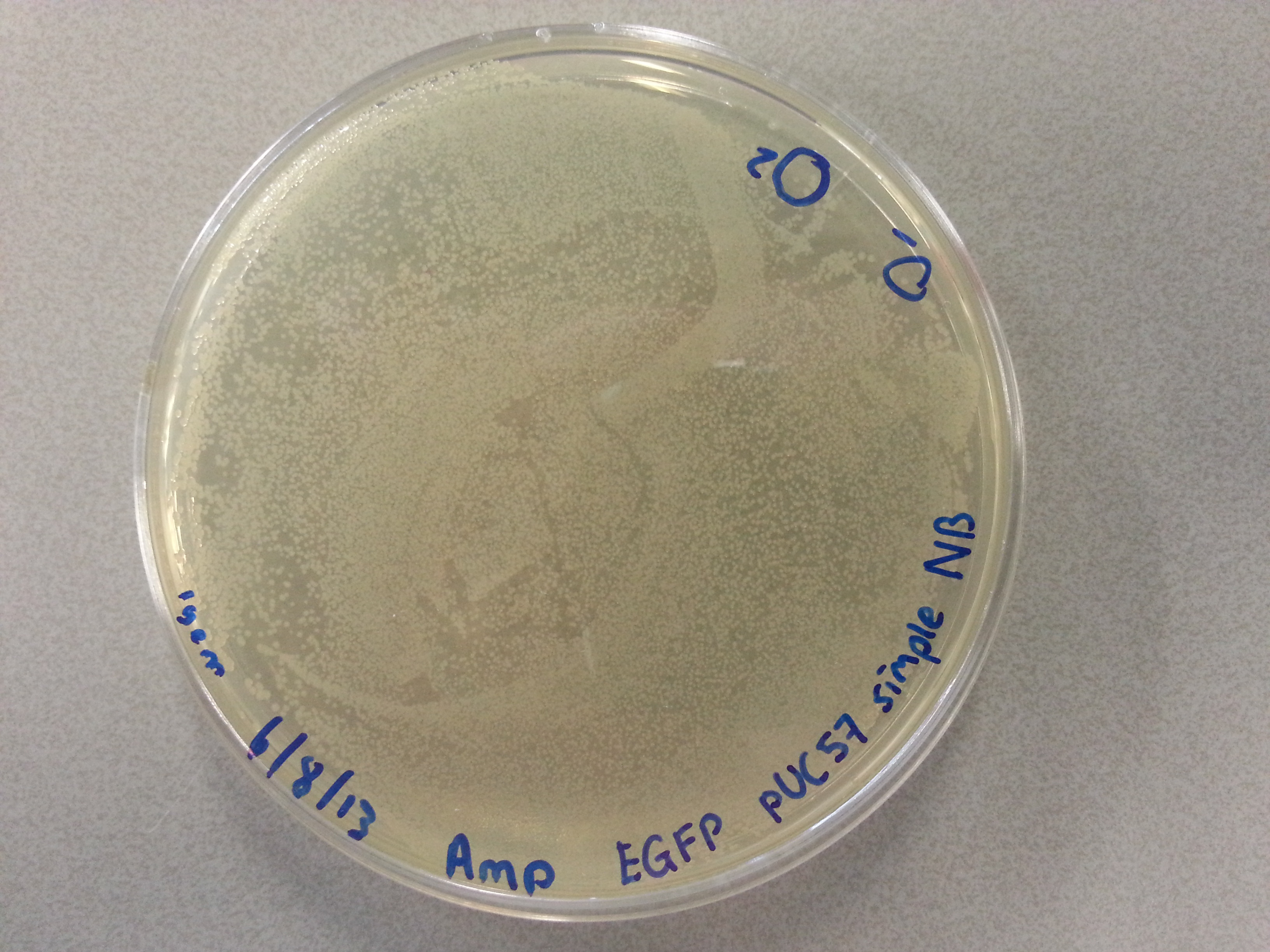
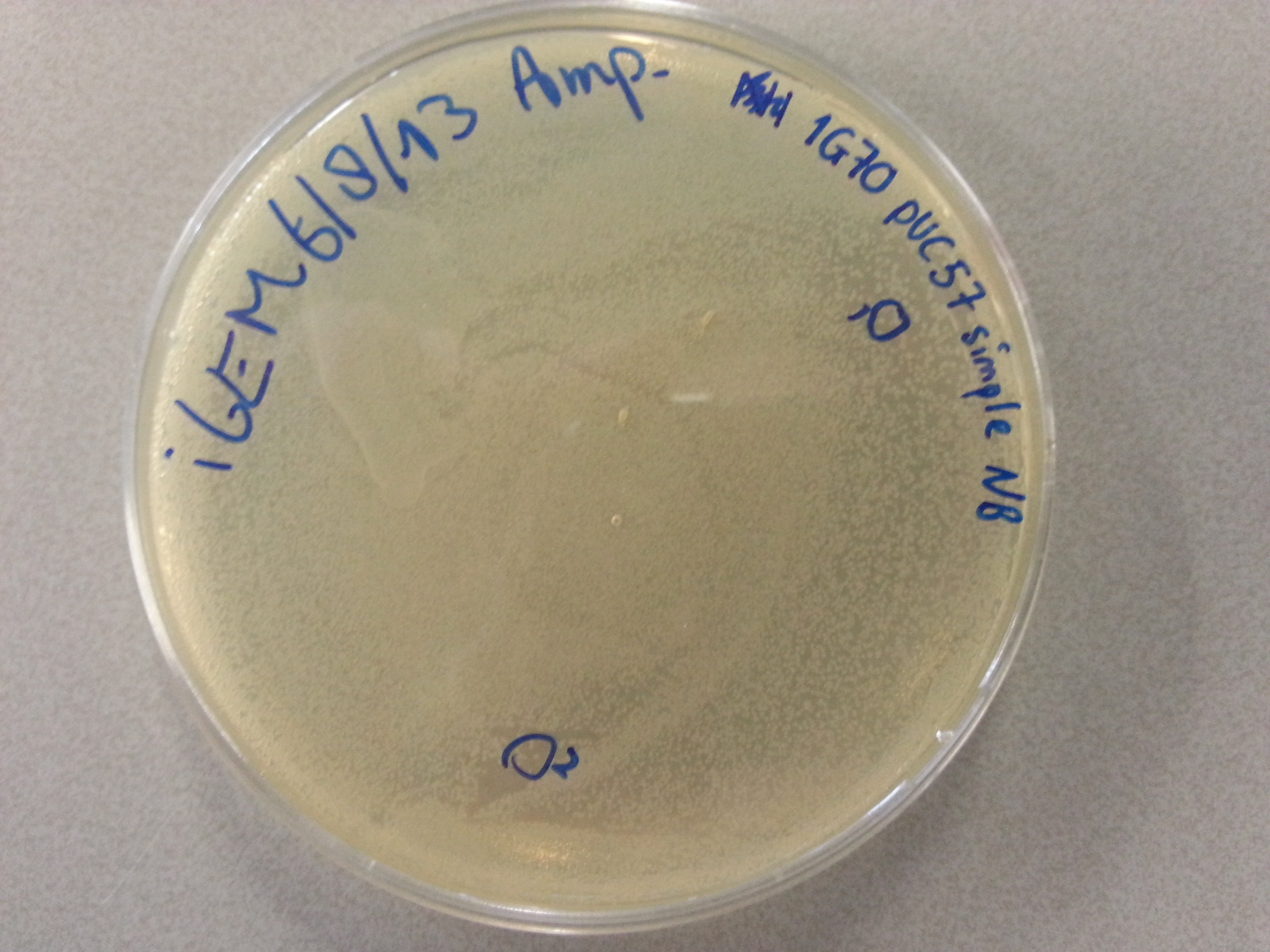
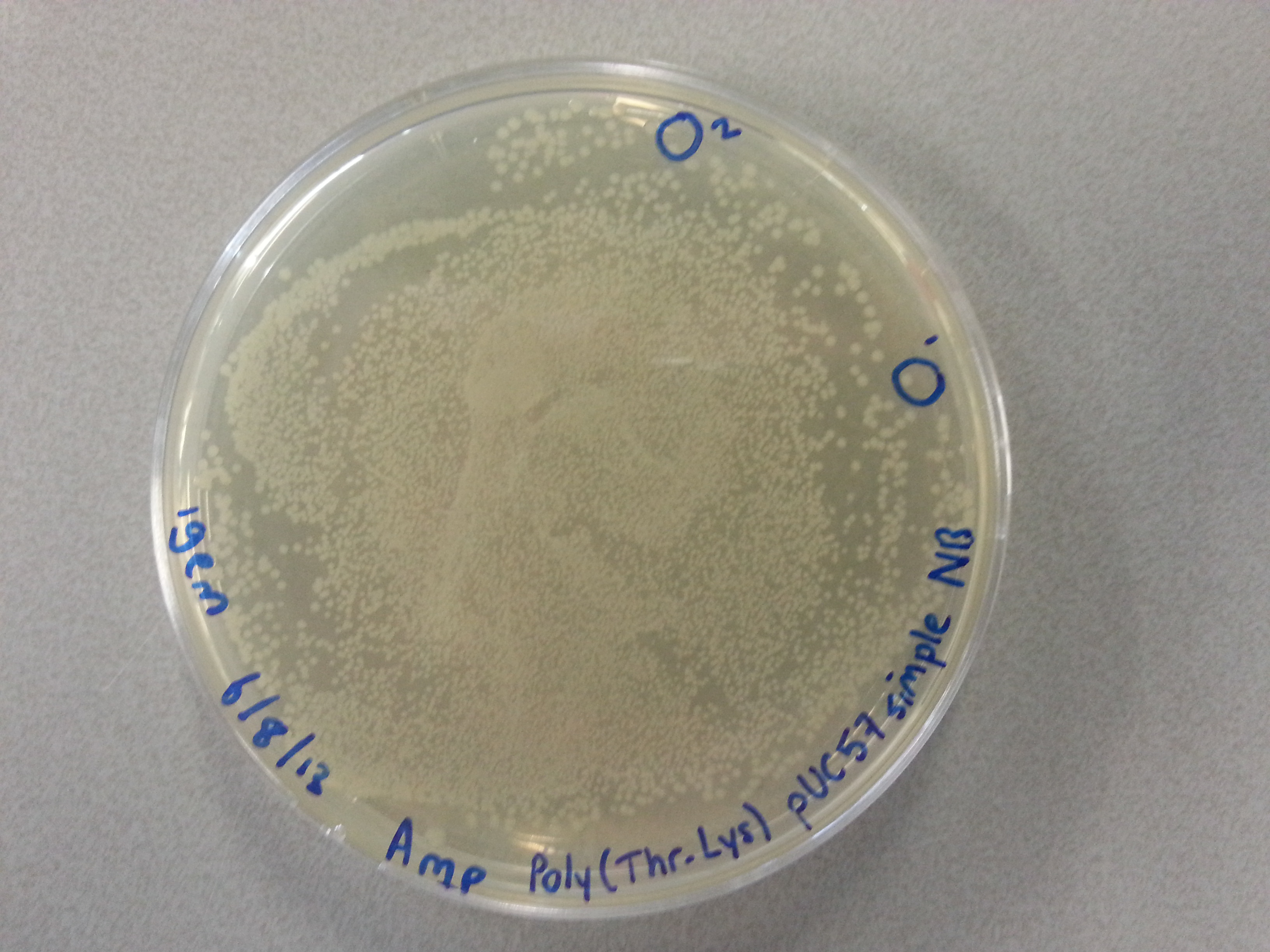
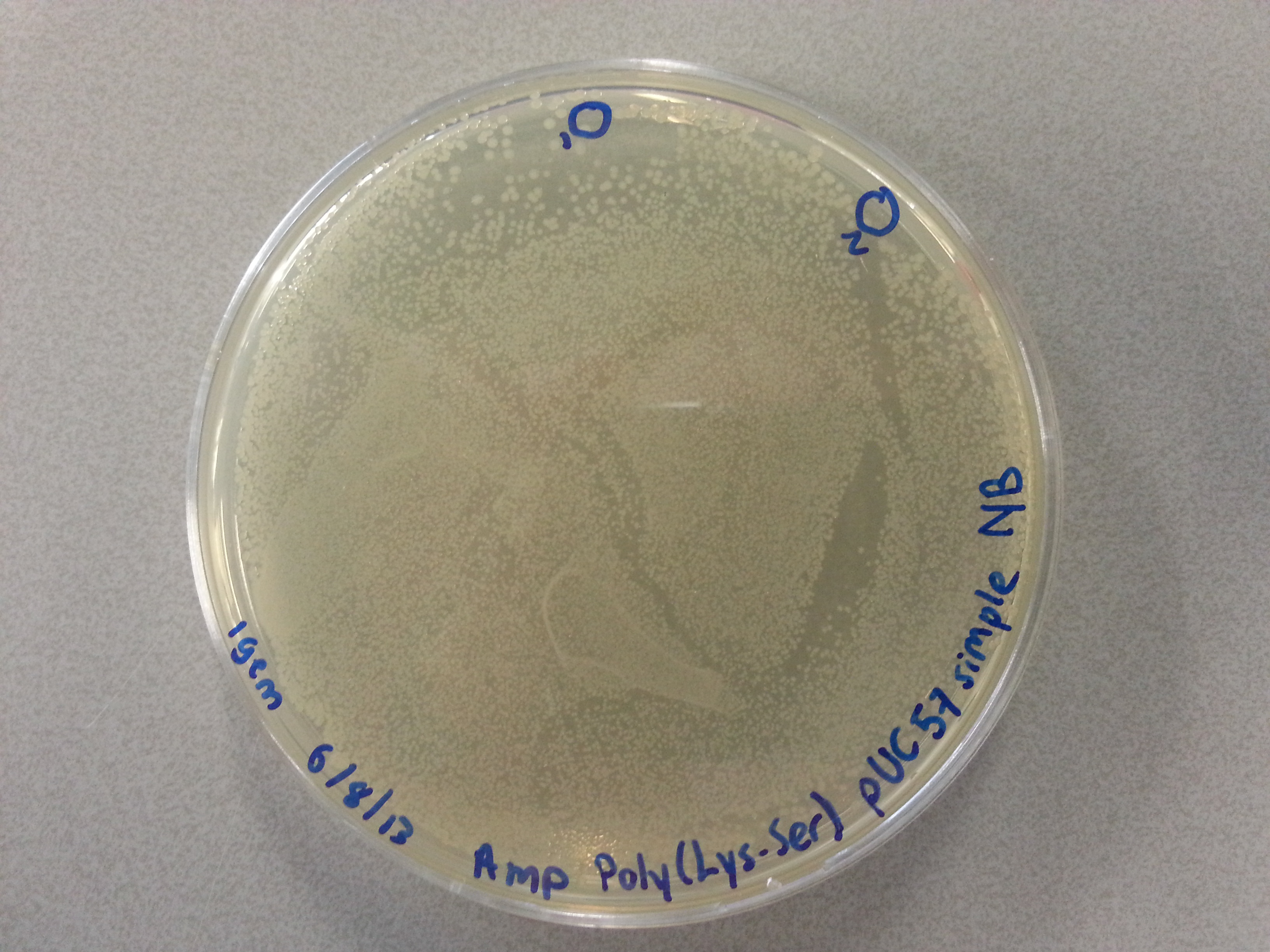
- 7.5µL of this bacterial picking solution was then pipetted into one of the culture tubes, until all tubes contained a portion of bacteria. Great care was taken in the labelling of the cultures so we could trace back which picking had been inserted into which tube.
- The culture tubes were then placed in the 37°C incubator and left for overnight growth.
It should be noted that four culture tubes were created per sample to ensure a large enough DNA amplification to be able to perform a restriction, gel extraction and ligation later on. Previously we had made 12 of these cultures per construct but it turned out that that was far too much (a miscalculation on our part), therefore we chose to create only four 8mL cultures per construct.
Culturing Old Constructs
Apart from just transforming and plating the new constructs mentioned above, we had also transformed and plated the older pET28a ligated constructs on August 12th. These too had been left to grow overnight and we were now ready to continue work on these samples. Photos are given here:
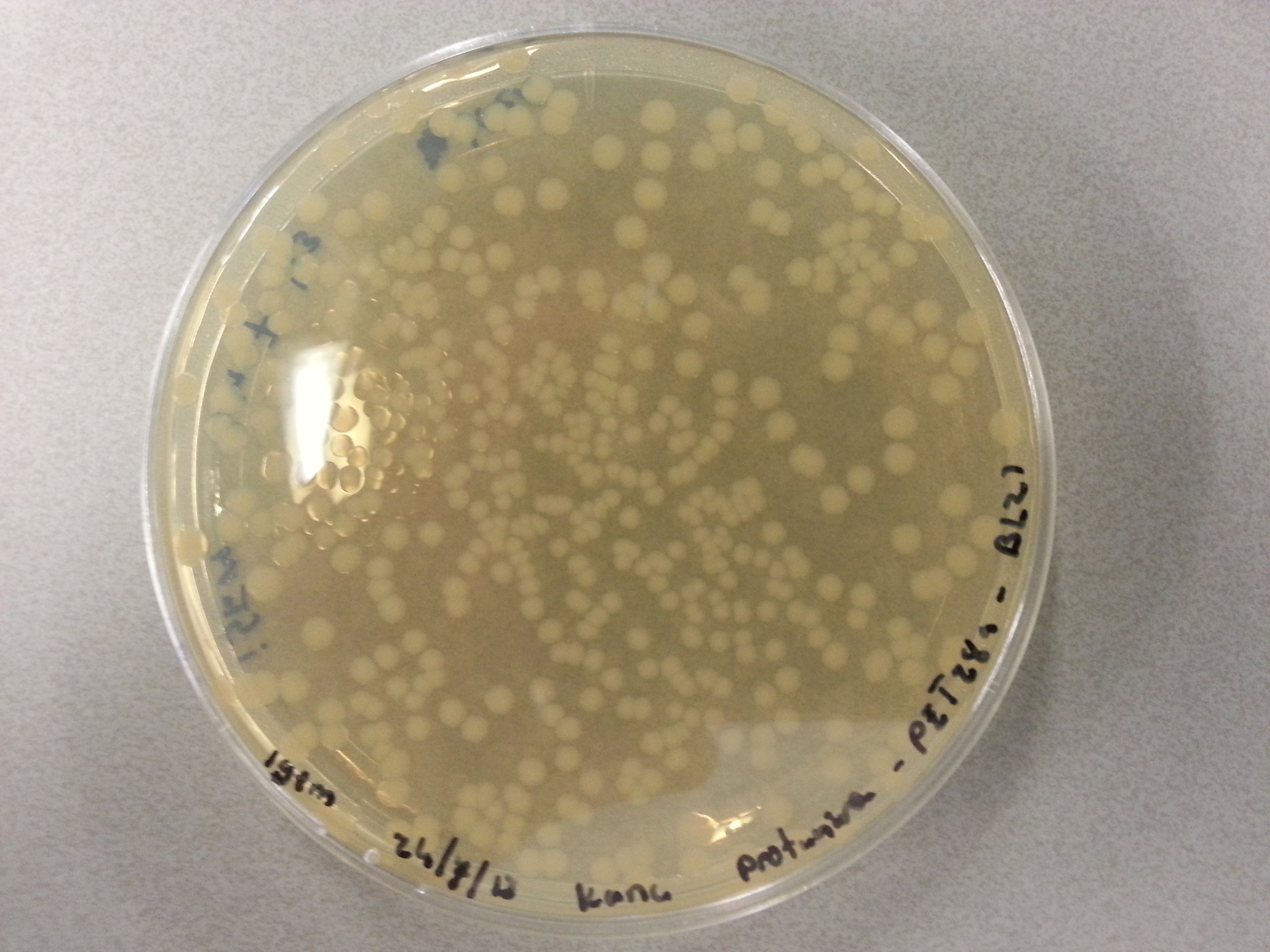
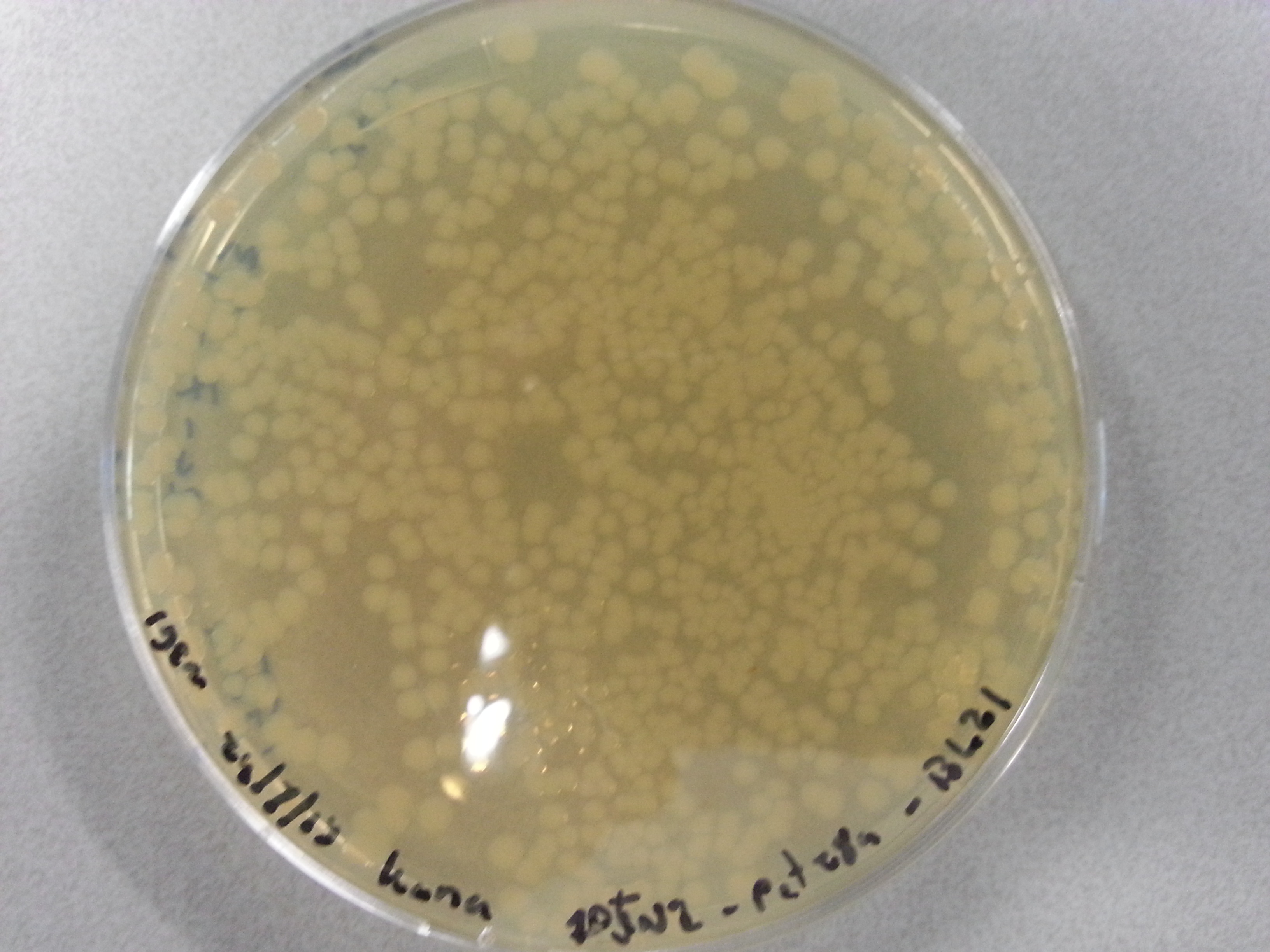
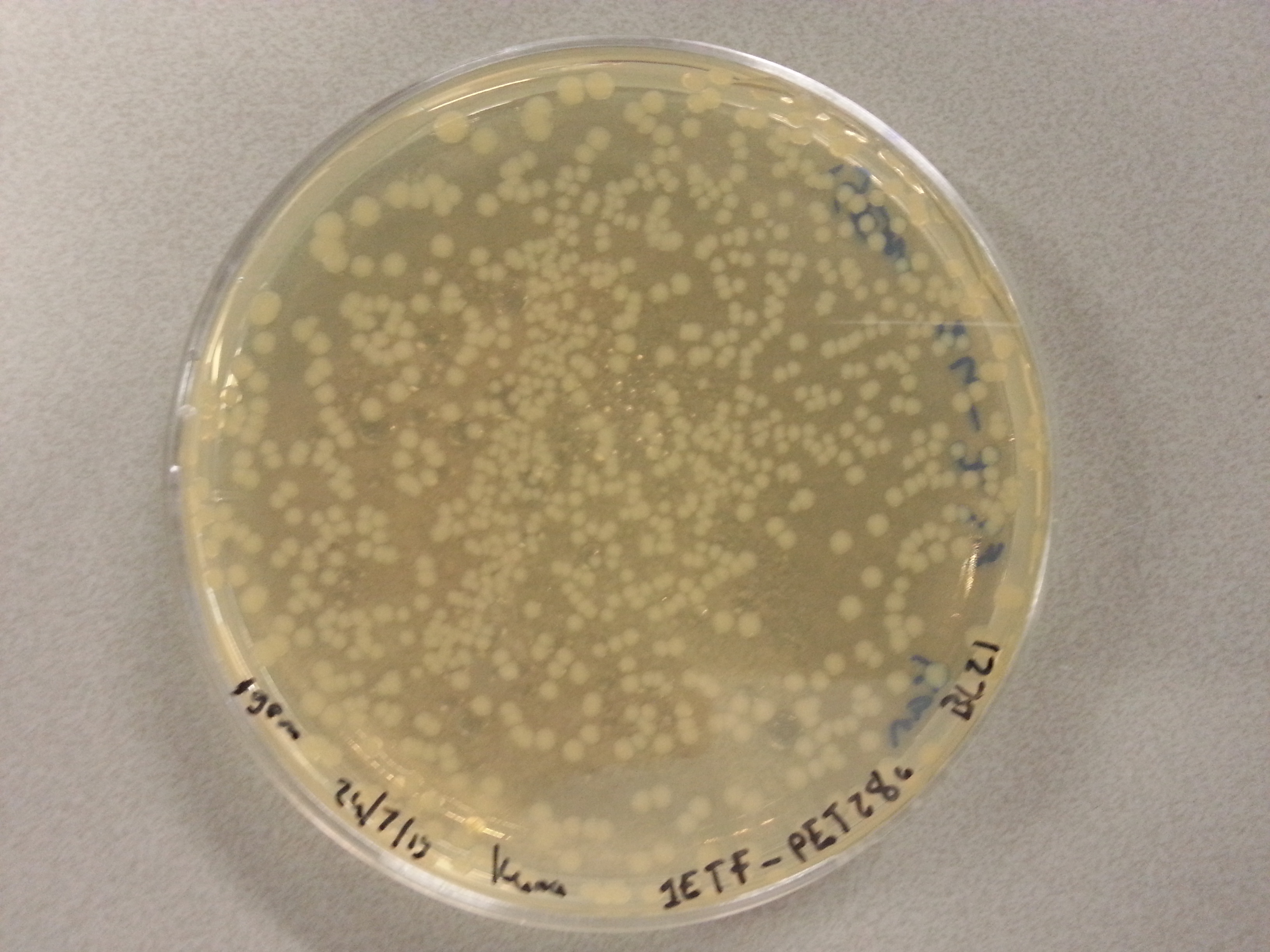
- Firstly two 100mL culture mediums were created by mixing the following in two small Erlenmeyer flask:
- 1g Peptones
- 1g NaCl
- 0.5g Yeast Extract
- 100mL Sterile Water
- The above mentioned solutions were then placed in the autoclave to be sterilised.
- After having cooled down 100µL of Kanamycin antibiotics were added to each of the flasks.
- Subsequently a single colony was picked from each of the following agar plates: Protamine-1-Optimized-pET28a and Poly(Arginine-Glycine)-pET28a. The two colonies were then transferred to one of the two flasks by ejecting the pipet point into the medium.
- The culture flasks were then placed in the 37°C incubator and left there to grow overnight.
Miniprepping New Constructs
On August 13th the four newest constructs (EGFP, 1G70, Poly(Threonine-Lysine) and Poly(Lysine-Serine)) were being cultured and were therefore placen in a 37°C incubator overnight. It was now time to retain the increased DNA using the miniprep protocol:
- First the culture tubes were spun down in a centrifuge for 10 minutes at 3700G causing the bacterial cells with the DNA inside to form pellets isolating it from the growth medium.
- The supernatant that had formed above the pellets was discarded and the pellets themselves were resuspended in 250µL of P1 buffer. The tubes were gently shaken by hand until the entire pellet had resuspended.
- The suspension was then transferred by means of pipetting into a smaller (1.5mL) eppendorf tube.
- 250µL of P2 buffer was then added to the eppendorf tube and the solution was inverted by hand until it had turned a clear blue.
- Within 5 minutes of adding the P2 buffer 350µL of N3 buffer was added to the solution and the eppendorf tube was inverted again until the solution was clear again.
- Once clear the eppendorf tube was placed in the centrifuge and spun for 10 minutes at 13000rpm seperating the bacterial cells from the DNA which remained suspended in the solution. The bacterial cells formed a pellet in the bottom of the eppendorf tube. (Unfortunately the pellet had not completely formed after 10 minutes so the eppendorf tube was spun for a further minute, again at 13000rpm.)
- As soon as the centrifuge had stopped and we could see that the pellet had properly formed, the supernatant was poured off into a special QIAcolumn. The pellet could be discarded in the bio-hazard waste.
- The QIAcolumn was subsequently centrifuged for one minute at 13000rpm, during which the DNA vector bound to the column meaning the flow through could be discarded.
- 750µL of PE buffer was then added to the QIAcolumn before centrifuging it for another minute at 13000rpm. The flow through was discarded.
- To ensure that all the PE buffer had passed through the column it was then spun yet again for one minute at a speed of 13000rpm. Yet again the flow through could be discarded.
- The column itself was then placed above a sealable 1.5mL eppendorf tube so that the DNA could be contained and stored. To obtain the DNA from the column 42µL of MilliQ water was pipetted onto the very center membrane of the column after which it was spun down for one minute at 13000rpm. The flow through now sat in the 1.5mL eppendorf tube and contained the DNA vectors.
The following concentrations were then measured for each of the DNA samples:
| Vector | Concentration (ng/µL) |
|---|---|
| EGFP | 208.9 |
| 1G70 | 165.6 |
| Poly(Threonine-Lysine) | 139.7 |
| Poly(Lysine-Serine) | 136.5 |
Digestion
As we did for the previous batch of constructs we would now digest these newest constructs. After digestion we would then, as we did previously for the older constructs have to perform gel extraction to purify the digestion products of these constructs. This would mean digesting a total of 20µg of each construct. This would provide us with 10µg to digest for ligation into the pET28a vector and 10µg of to digest for ligation into the pBR322 vector. For the former digestion the XhoI and NheI restriction enzymes would be used. For the latter digestion we would use the EcoRI and HindIII restriction enxymes, to later allow us to ligate the restriction product into the pBR322 vector. As digestion is not optimal when 10µ of DNA are digested in a single solution each construct would be digested in four separate solutions each containing 5µg. Two of these solutions would contain the enzymes for ligation into the pET28a vector and the remaining 2 solutions the enzymes for ligation in pBR322. The Complete solution for the digestions are given below:
| Construct | Concentration (ng/µL) | Volume for 5µg of DNA (µL) | Cutsmart Buffer (µL) | Enzyme (µL) | Sterile Water (µL) |
|---|---|---|---|---|---|
| EGFP | 208.9 | 23.9 | 5.0 | 0.7 | 19.7 |
| 1G70 | 165.6 | 30.2 | 5.0 | 0.7 | 13.4 |
| Poly(Threonine-Lysine) | 139.7 | 35.8 | 5.0 | 0.7 | 7.8 |
| Poly(Lysine-Serine) | 136.5 | 36.6 | 5.0 | 0.7 | 7.0 |
As mentioned two restriction enzymes were added to each solution. The given Enzyme volume is the amount that should be added of each of the two enzymes. It should also be noted that for the Poly(Threonine-Lysine) and Poly(Lysine-Serine) there was a slight deficit in the amount of DNA available for one of the solutions. This meant that one solution did not contain 5µg of DNA but slightly less (approx. 4.5µg).
We also had to digest some more of both the vectors. For each vector we aimed to digest a total of 2µg. Therefore the following solutions were made:
| Vector | Volume for 5µg of DNA (µL) | Cutsmart Buffer (µL) | Enzyme (µL) | Sterile Water (µL) |
|---|---|---|---|---|
| pET28a | 18.5 | 5.0 | 0.7 | 25.8 |
| pBR322 | 32.2 | 5.0 | 0.7 | 12.1 |
Here we used the vector DNA that was prepared in the earlier stages (see August 2nd).
These solutions (Both for the vectors as well as the constructs were then placed in the PCR apparatus and set witht he following time schedule:
- Reaction step: 1 hour at 37°C
- Enzyme inactivation step: 20 minutes at 80°C
- Storage step: Forever at 4°C
The restriction products were then run and left at 4°C until they were needed again the next day.
Testing the Expression
On August 13th 2 cell cultures had been placed in the 37°C incubator to grow the bacteria overnight. It was now time to induce the expression. This would be done aerobically for the time being. It is important to test the protein expression on a small scale to start with so that you reduce risks when scaling up. It was decided that once we had induced expression by addition of IPTG that we would take samples of the expression medium every hour to get a good view of the expression curve. These samples would be collected today and analysed tomorrow. The following procedure was followed in order to obtain our samples:
- An Optical density measurement of the cell culture was taken to check the cell growth. An optical density above 0.6 would mean that expression induction would be possible. The following optical densities were found:
- Protamine culture: 2.140
- Poly(Arginine-Glycine) culture: 2.287
- For this reason 50µL of IPTG was added to each of the cultures. After having placed each culture in the incubator for a further 15 minutes it occurred to us that an optical density above 2 might be far to high for proper expression as the medium would not contain large enough food supplies. The following emergency procedure was then performed with haste:
- First three test sample tubes were made each with a different amount of new LB medium and a small amount of our existing cell culture:
- 11mL LB and 1mL cell culture
- 10mL LB and 2mL cell culture
- 8mL LB and 4mL cell culture
- These three test tubes were then measured for their optical density. It was revealed that a dilution containing 10mL of LB medium and 2mL of our existing cell culture was optimal. Therefore 2 new culture tubes were filled with 10mL's of LB medium. To each we then added 2.5mL of one of the two cell cultures. We also added 12.5µg of Kanamycin antibiotics and an extra 5µL of IPTG to account for the dilution factor. The optical densities were then measured of these new culture tubes:
- Protamine cell culture: 0.50
- Poly(Arginine-Glycine) cell culture: 0.56
- These Optical densities were adequate and therefore a t=0 sample was collected by pipetting 1.5mL of the cell culture into an eppendorf tube which was subsequently placed in the 4°C refrigerator.
- First three test sample tubes were made each with a different amount of new LB medium and a small amount of our existing cell culture:
- The new cell cultures were then placed in the 37°C incubator and 1.5mL samples of each were taken every hour from then on for a further 5 hours. Each sample was immediately placed in the 4°C refrigerator.
Preparing an SDS-gel
Tomorrow we would like to run a number of samples on SDS gel to see whether the protein expression had been successful. In preparation of this we prepared two 12% SDS gels. To start with two solutions were prepared, a running and a stacking gel solution. The recipes for both are given below:
| 12% Running Gel (mL) | Stacking Gel (mL) | |
|---|---|---|
| Sterile Water | 9.9 | 5.5 |
| 30% Acrylamide Mix | 12 | 13 |
| 1.5M Tris-HCl Buffer (pH 8.8) | 7.5 | 0 |
| 1M Tris-HCl Buffer (pH 6.8) | 0 | 1 |
| 10% SDS | 0.3 | 0.08 |
| 10% Ammonium Persulfate | 0.3 | 0.08 |
| TEMED | 0.012 | 0.008 |
It should be remembered that the 10% Ammonium Persulfate and the TEMED should only be added once ready to pour, these will ensure the solutions harden into gels.
Having made the above solutions they were poured between too securely held glass plates. First the 12% running gel was poured and topped off with water to level it out. Once hard, the water could be poured off and the stacking gel could be added and allowed to hardened. Once hard the gels were wrapped in damp tissues and then aluminium foil. The entire package was then placed in the 4°C refrigerator overnight for use tomorrow.
Running the SDS-Gel
On August 14th an SDS gel was prepared so that we would be able to run our expression samples today. Before being able to load the expression samples these first had to be prepared:
- We had taken 6 1.5mL samples for each of the two expression cultures (Protamine-1-Optimized and Poly(Arginine-Glycine)). For each of the samples we wished to isolate the bacteria containing our proteins from the LB medium. This was done by centrifuging the samples at 13400 rpm in a tabletop centrifuge for 8 minutes.
- During the centrifuging a pellet had formed in the bottom of the eppendorf tube. This contained the bacteria and therefore also our proteins. The supernatant could therefore be discarded.
- To isolate our proteins from the bacteria we would have to add both BugBuster and Benzonse. For BugBuster 2mL was added for every 100mL of culture we wished to re-suspend. For Benzonase we would have to add 1/1000th of the volume of the BugBuster that we added. As this would be a very small amount for each of the samples separately a master mix was made by mixing 500µL of Bugbuster with 0.5µL of Benzonase.
- Of this master mix 30*micro;L was added to each of the pellets. These were then placed in a tabletop shaker for 25-30 minutes at 25°C to resuspend the pellet.
- Once resuspended the solution was spun down again for 5 minutes at 13400 rpm in a tabletop centrifuge. Once more a pellet of bacteria formed only this time the proteins could be found in the supernatant. A portion of this supernatant would therefore be loaded onto the SDS gel. However it was also possible that the proteins we wanted to see remained in the inclusion bodies in the bacteria, therefore we would also have to run these on gel.
- 15µL of the supernatant was pipetted into a separate eppendorf tube ready for loading. The rest of the supernatant was then discarded.
- !00µL of pH 8.8 buffer was then added to the pellet which resuspended. Of this solution 15µL was also pipetted into a separate eppendorf tube also ready for gel loading.
- For each of the two cultures we now had 12 samples. six of these were supernatant samples containing primarily proteins, the other six were solutions containing dissolved bacterial pellets. Each sample tube contained 15µL of solution.
- TO be able to see the samples on the gel a loading dye would have to be added to each sample. The loading dye was prepared by combining the following:
- 500µL of Loading Dye
- 1µL of DTT
- This created a 2x loading buffer that could be added to all the samples.
- 15µL of the loading dye solution was then added to each of the samples which were subsequently ready for loading.
- 11µL of each sample was loaded into the separate columns of the 12% SDS gel which was subsequently run with the following program:
- 15 minutes at 90V for the stacking equality.
- 40 minutes at 150V through the running gel.
- After running the gels they were shortly washed with water three times before being placed in a water bucket on the shaking table for 15 minutes.
- Hereafter the water was removed and the gels were paced in Coomassie Blue staining medium for two hours.
- The gels were then returned to water and left on the shaking table overnight.
Digestion Purification
On August 14th the new construct were digested along with the vecotrs pBR322 and pET28a. The constructs would have to be purified by performing a gel extraction whereas the vectors could be purified simply by performing the PCR purification protocol.
Purifying the Vectors
To purify the vectors after digestion, in preparation of the ligation the following steps were followed:
- To the PCR product 5 volumes of PB buffer were added (In our case this was 250µL of PB buffer as we had 50µL of PRC product.)
- This solution was placed in a QIAquick spin column and spun for 60 seconds at 13000 rpm. The flow through could be discarded.
- Now 750µL of PE buffer could be added to the column before spinning it down once more for 60 seconds at 13000 rpm.
- Having discarded the flow through the column was spun for a further 60 seconds again at 13000 rpm to remove the last of the PE buffer.
- The column was then placed into a sealable eppendorf tube before adding 42µL of MilliQ water to the very centre membrane of the spin column and leaving it to stand for one minute.
- The column was now placed in the centrifuge and spun for 60 seconds at 13000 rpm producing a solution containing the desired DNA.
Purifying the Inserts
The inserts too would need to be purified however as they were quite large this would not be possible using the PCR purification kit. This meant that we would have to load and extract the digestion products on an agarose gel. For each of the four constructs (EGFP, 1G70, Poly(Threonine-Lysine), Poly(Lysine-Serine)) we had four PCR products. Two of these had been digested for ligation into a pET28a vector and the other two solutions had been digested for ligation into pBR322.
- The first step was the combination of the two solutions per construct which had been digested for ligation into the same vector.
- After combination a total volume of 100µL had been attained. Therefore 20µL of 6x Loading Dye was added to each solution.
- This entire solution was then loaded into a single column on a 1% agarose gel prepared by mixing the following:
- 1.2g of Agarose
- 120mL of TAE buffer
- 12µL of CyberSafe
- These gels were poured and let to harden before the samples were loaded into specially broadened columns.
- Once fully loaded the gel was run at 100V for an hour.
After running the gel, gel extraction could be performed in the following manner:
- First the gels were placed on a blue light to light up the bands and the correct bands were cut out. (These were the lower bands as the inserts had a smaller base length than the construct they were restricted out of).
- A gel extraction column can hold a maximum of 400mg of gel. The gel cut-outs all weighed more than this and therefore each of the bands were split into two halves. Each half was placed in a separate 2mL eppendorf tube and weighed.
- To each tube 3 volumes of QG buffer was added. A volume can be defined as the weight of the gel in mg transferred into µL. So a gel weighing 400mg would have a volume of 400µL, so 1200µL of QG would be added.
- The eppendorf tubes containing the gel cut-outs and the QG buffer were placed in an incubator set at 50°C for 10 minutes, whilst vortexing the samples at regular (every 2 minutes) intervals until the gel had completely dissolved.
- To these tubes 1 volume of isopropanol was added.
- Each tube was then brought over into a QIAquick column and placed in the centrifuge where it was spun down for 60 seconds at 13000 rpm. The flow through was then discarded. If the volume of the gel, QG buffer and Isopropanol was larger than 800µL then this step was performed twice in the same column.
- Now 750µL of PE buffer was added to the column before centrifuging at 13000 rpm for 60 seconds. After discarding the flow through the same amount was added and once more and again centrifuged.
- Removing the flow through the column was then centrifuged again for 60 seconds at 13000 rpm removing the last of the PE buffer.
- After bringing the column across into a sealable eppendorf tube 36µL of MilliQ water was pipetted onto the very centre membrane of the column which was then let to stand for 60 seconds. Subsequently the column was spun down at 13000 rpm for 60 seconds. The DNA is now in the flow through.
- Remembering that we split the gel in half in the very first stage of this protocol, the two solutions containing the same DNA were combined so that a total volume of maximally 72µL of each insert was formed.
Nanodrop Results
For the ligation the concentrations of both the digested vectors and the digested inserts would have to be measured. The results can be seen in the tables below. The first table gives the concentration of both the vectors. The second gives the results for the digestion for ligation into pET28a and the third the results for ligation into pBR322
| Vector | Concentration (ng/µL) |
|---|---|
| pET28a | 13.2 |
| pBR322 | 5.2 |
| pET28a Digested Inserts | Concentration (ng/µL) |
|---|---|
| EGFP | 17.6 |
| 1G70 | 8.8 |
| Poly(Threonine-Lysine) | 9.5 |
| Poly(Lysine-Serine) | 11.5 |
| pBR322 Digested Insert | Concentration (ng/µL) |
|---|---|
| EGFP | 9.6 |
| 1G70 | 6.0 |
| Poly(Threonine-Lysine) | 3.9 |
| Poly(Lysine-Serine) | 3.2 |
As we can see the digestion was unsuccessful for the pBR322 vector. Its concentration is too low to be of use for the ligation which we performed next. Luckily however we had an older pBR322 digested sample in the freezer with a concentration of 13.1ng/µL which we could use.
Ligation
Having determined the concentrations of each of the constructs and the vectors we could start performing the calculations for the ligations. We wished to ligated 100ng of each of the vectors with each of the accordingly digested inserts. Ligation works best when there is more insert available than there is vector. Therefore we would first need to find out how many ng of insert will be needed for molar equality. This is done by dividing 100ng (the amount of vector we are using) by the number of bases in the vector. This amount would then be multiplied by the number of bases in the insert to reveal the mass in ng of insert we would need. Using this and with the concentrations of each insert at hand we could then determine the volume of DNA we would need to add to obtain a molar equality. Multiplying this volume by five would ensure a multitude of insert relative to the number of vectors in the ligation solutions.
Here the volume of each vector solution needed for 100ng of DNA:
| Vector | Concentration (ng/µL) | 100g DNA (µL) | Number of Bases in Vector |
|---|---|---|---|
| pBR322 | 13.1 | 7.6 | 4330 |
| pET28a | 13.2 | 7.6 | 5296 |
The following table gives the volumes needed for 5x molar equality of each of the inserts for the pET28a vector:
| Construct | Number of Bases | Concentration (ng/µL) | ng Needed for Molar Equality (ng) | Volume Needed for Molar Equality (µL) | Volume Needed for 5x Molar Equality (µL) |
|---|---|---|---|---|---|
| EGFP | 732 | 9.6 | 13.8 | 1.44 | 7.1 |
| 1G70 | 417 | 6.0 | 12.7 | 2.12 | 10.6 |
| Poly(Threonine-Lysine) | 447 | 3.9 | 8.4 | 2.15 | 10.8 |
| Poly(Lysine-Serine) | 447 | 3.2 | 8.4 | 2.63 | 13.1 |
The following table gives the volumes needed for 5x molar equality of each of the inserts for the pBR322 vector:
| Construct | Number of Bases | Concentration (ng/µL) | ng Needed for Molar Equality (ng) | Volume Needed for Molar Equality (µL) | Volume Needed for 5x Molar Equality (µL) |
|---|---|---|---|---|---|
| EGFP | 1056 | 9.6 | 13.8 | 1.44 | 7.1 |
| 1G70 | 741 | 8.8 | 17.1 | 1.94 | 9.7 |
| Poly(Threonine-Lysine) | 771 | 9.5 | 17.8 | 1.87 | 9.4 |
| Poly(Lysine-Serine) | 771 | 11.5 | 17.8 | 1.55 | 7.7 |
Using the above tables we now had all the information to be able to prepare our ligation solutions:
| pET28a Ligation Solutions | Vector Volume (µL) | Insert Volume (µL) | 10x Buffer (µL) | Sterile Water (µL) | Ligase (µL) |
|---|---|---|---|---|---|
| EGFP | 7.6 | 7.1 | 2.5 | 6.8 | 1 |
| 1G70 | 7.6 | 10.6 | 2.5 | 3.3 | 1 |
| Poly(Threonine-Lysine) | 7.6 | 10.8 | 2.5 | 3.1 | 1 |
| Poly(Lysine-Serine) | 7.6 | 13.1 | 2.5 | 0.8 | 1 |
| pBR322 Ligation Solutions | Vector Volume (µL) | Insert Volume (µL) | 10x Buffer (µL) | Sterile Water (µL) | Ligase (µL) |
|---|---|---|---|---|---|
| EGFP | 7.6 | 6.1 | 2.5 | 7.0 | 1 |
| 1G70 | 7.6 | 9.7 | 2.5 | 4.2 | 1 |
| Poly(Threonine-Lysine) | 7.6 | 9.4 | 2.5 | 4.5 | 1 |
| Poly(Lysine-Serine) | 7.6 | 7.7 | 2.5 | 7.2 | 1 |
Having prepared the above 25µL ligation solutions they were placed in the PCR apparatus with the following program:
- Ligation: 2 hours at 25°C
- Denaturation: 10 minutes at 65°C
- Storage: FOREVER at 4°C
The ligation products were then left in the PCR apparatus at 4°C overnight ready for transformation and plating the following day.
Transforming the Ligation Products
After having ligated our four newest constructs on August 15th into both pET28a and pBR322 we were now able to transform these into NB ready for growth on an agar plate.
- First 1µL of the ligation product was pipetted into 20µL of NB bacteria. These were then left to stand on ice for 5 minutes.
- The heat shock was performed by transferring the solutions to a 42°C water bath for 30 seconds before returning to ice for a further two minutes.
- Then 80µL of SOC solution was added to the bacteria where after they were not returned to ice.
- The transformation solutions were then incubated for 60 minutes at 37°C.
Plating
To grow cultures containing our ligation products we plated our transformation solutions. The constructs ligated into pET28a were plated on Kanamycin agar plates and those ligated into pBR322 were plated onto Ampicillin agar plates. This was done as follows:
- All steps were performed in the vicinity of a blue bunsen flame.
- Each transformation solution was pipetted in its entirety onto one of the plates.
- Using a sterile spreader the solution was spread out over the plate.
- The plates were then placed in a 37°C incubator overnight. As it was a Friday the incubator was set to 4°C for the remainder of the weekend after the plates had grown or 15 hours.
Week 5
Colony Picking and PCR
Previously, on August 16th, agar plates had been prepared by transforming the ligations of the four newest constructs (EGFP, 1G70, Poly(Threonine-Lysine) and Poly(Lysine-Serine)) in both the pET28a and pBR322 vectors into NovaBlue bacteria and plating this entirety onto either Ampicillin or Kanamycin agar plates, according to the vector (Ampicillin plates for pBR322 ligated vectors and Kanamycin plates for pET28a ligated vectors.) Having grown these plates overnight, and storing them at 4°C for the remainder of the weekend we were now able to perform colony picking and PCR. The images of the plates are given here:
- 216µL of sterile water.
- 300µL of Kapa 2G.
- 30µL of the according FWD primer.
- 30µL of the according RW primer.
Having prepared the two master mixes, 20 PCR eppendorf tubes per master mix were filled with 24µL of that master mix solution. To each of these prepared eppendorf tubes 1µL of the culture picking dilution was added, making care that the eppendorf tubes were labelled in the same manner as before and that the pBR322 ligated constructs were placed in the master mix containing the primers for that vector. The same was then done for the pET28a ligated constructs. Having prepared these samples they were placed in the PCR apparatus and set with the following protocol:
- Initial Denaturation: 95°C for 3 minutes.
- The following steps should be performed in this order, repeating the process a total of 34 times.
- Denaturation: 95°C for 15 seconds.
- Annealing: 58°C for 15 seconds.
- Extension: 72°C for 20 seconds.
- Final Extension: 72°C for 10 minutes.
- Storage: 4°C FOREVER.
Having completed the PCR protocol 4µL of 6x loading dye was added to each of the samples. From this solution now 18µL was loaded onto a 1.5% agarose gel which was prepared by mixing the following:
- 1.8g of Agarose
- 120mL of TAE buffer
- 12µL of Cybersafe
This gel was then run at 100V for 60 minutes.
Small cultures
Having run the gel with the colony PCR products we were able to analyse this and see in which of our colony pickings the ligated had been successful. For each of the ligated constructs, two successfully ligated pickings were then chosen for cell culture allowing us to multiply the DNA we had. This was done by adding 5µL of the original cell picking dissolved in water to an 8mL LB culture medium. To this culture medium we then also added 8µL of Ampicillin or Kanamycin dependant on the vector of the construct (Ampicillin for pBR322 ligated structures and Kanamycin for pET28a ligated structurs). These small culture tubes were then placed in a 37°C incubator for overnight growth.
Preparation for Protein Expression
Our previous test of protein expression had not been quite as successful as we had hoped as we had seen on August 16th. Therefore we were going to try again using two other constructs this time round namely: 1PJN and 1ETF both ligated into the pET28a vector. Each of these had already been transformed into BL21 bacteria and plated onto agar. We wished now to first grow these constructs on a small scale before transferring them to a larger culture for expression. To do this two culture tubes were prepared containing 10ml of LB and 10µL of Kanamycin. To each of these a pipet point was added with on the end a culture picking of one of the two constructs. These cultures were then placed in the 37°C incubator and left there for overnight growth. As we would be using the cell pellet formed after spinning down the expression product to measure the CEST contrast later this week, we would also need to provide a number of controls. One of these would be a cell pellet of a culture where no expression had taken place and a second would be the same only coated with poly-Lysines. To create the cell cultures for these controls 10mL of LB and 10µL of Kanamycin were mixed and to this solution a culture picking of 1ETF in BL21 bacteria was added. These too were cultured overnight at 37°C.
One further step was the preparation of two large cultures for the expression tomorrow. This was done by preparing 400mL of LB in two large erylmeyers. For the LB the following were mixed:
- 4g of Peptones.
- 4g of NaCl.
- 2g of Yeast Extract.
- 400mL of De-mineralised Water.
The solution was then autoclaved and left overnight ready for expression.
Miniprepping new constructs
On August 18th the four newest constructs (EGFP, 1G70, Poly(Threonine-Lysine) and Poly(Lysine-Serine)) had been ligated into both pBR322 and pET28a vectors. On August 19th a number of samples of these ligation products were taken and cultured in 8mL culture tubes. Today we would isolate the DNA vectors from the culture medium, send off DNA for sequencing and ultimately transform the DNA to BL21 bacteria which could be plated. To start off DNA would have to be retained which meant miniprepping the cultures, the protocol for which is given here:
- Firstly 700µL of each culture was pipetted into a storage tube along with 300µL of 50% Glycerol solution. The entire solution was then snap frozen in liquid nitrogen so that it could be stored in the -80°C freezer should we ever need to continue work with these vectors.
- Now the culture tubes were spun down in a centrifuge for 10 minutes at 3700G causing the bacterial cells with the DNA inside to form pellets isolating it from the growth medium.
- The supernatant that had formed above the pellets was discarded and the pellets themselves were resuspended in 250µL of P1 buffer. The tubes were gently shaken by hand until the entire pellet had resuspended.
- The suspension was then transferred by means of pipetting into a smaller (1.5mL) eppendorf tube.
- 250µL of P2 buffer was then added to the eppendorf tube and the solution was inverted by hand until it had turned a clear blue.
- Within 5 minutes of adding the P2 buffer 350µL of N3 buffer was added to the solution and the eppendorf tube was inverted again until the solution was clear again.
- Once clear the eppendorf tube was placed in the centrifuge and spun for 10 minutes at 13000rpm seperating the bacterial cells from the DNA which remained suspended in the solution. The bacterial cells formed a pellet in the bottom of the eppendorf tube. (Unfortunately the pellet had not completely formed after 10 minutes so the eppendorf tube was spun for a further minute, again at 13000rpm.)
- As soon as the centrifuge had stopped and we could see that the pellet had properly formed, the supernatant was poured off into a special QIAcolumn. The pellet could be discarded in the bio-hazard waste.
- The QIAcolumn was subsequently centrifuged for one minute at 13000rpm, during which the DNA vector bound to the column meaning the flow through could be discarded.
- 750µL of PE buffer was then added to the QIAcolumn before centrifuging it for another minute at 13000rpm. The flow through was discarded.
- To ensure that all the PE buffer had passed through the column it was then spun yet again for one minute at a speed of 13000rpm. Yet again the flow through could be discarded.
- The column itself was then placed above a sealable 1.5mL eppendorf tube so that the DNA could be contained and stored. To obtain the DNA from the column 42µL of MilliQ water was pipetted onto the very center membrane of the column after which it was spun down for one minute at 13000rpm. The flow through now sat in the 1.5mL eppendorf tube and contained the DNA vectors.
The following concentrations were then measured for each of the DNA samples:
| Construct | Ligation Vector | Colony Picking Number | Concentration (ng/µL) |
|---|---|---|---|
| EGFP | pET28a | 1 | 63.1 |
| EGFP | pET28a | 2 | 62.8 |
| EGFP | pBR322 | 1 | 71.0 |
| EGFP | pBR322 | 2 | 64.5 |
| 1G70 | pET28a | 1 | 63.4 |
| 1G70 | pET28a | 2 | 75.1 |
| 1G70 | pBR322 | 3 | 72.2 |
| 1G70 | pBR322 | 5 | 73.4 |
| Poly(Threonine-Lysine) | pET28a | 2 | 62.2 |
| Poly(Threonine-Lysine) | pET28a | 4 | 65.8 |
| Poly(Threonine-Lysine) | pBR322 | 2 | 49.5 |
| Poly(Threonine-Lysine) | pBR322 | 3 | 47.0 |
| Poly(Lysine-Serine) | pET28a | 1 | 61.9 |
| Poly(Lysine-Serine) | pET28a | 2 | 60.6 |
| Poly(Lysine-Serine) | pBR322 | 1 | 55.8 |
| Poly(Lysine-Serine) | pBR322 | 3 | 55.1 |
Sequencing
To check the sequences of our DNA vectors we again decided to send our vectors off to an external sequencing agency. For this they request a 7µL solution which contains 1µL of 10µM primer. They also request at least 400ng of DNA for successful sequencing. As the pBR322 ligated Poly(Threonine-Lysine) and Poly(Lysine-Serine) had such low concentrations these could not be sent off. Of the other constructs only a few would contain enough DNA but we would hope for the best. This meant that we would have to create two sample tubes per construct. One would contain the forward primer and one the reverse primer. We then added 6µL of each sample to the correct sample tube, being sure to label them well. Care should be taken to add the pET28a primers to the pET28a ligated constructs and the same of course for the pBR322 ligated constructs.
Transformation
From the above mentioned DNA samples we could now perform a transformation into BL21 bacteria. For this we would use the samples with the highest concentration from each pair of samples belonging to the same construct and ligation vector. (It should be noted that we only did this for the pET28a ligated constructs as these would all be expressed aerobically. We would not be bringing all constructs to expression anaerobically. However pBR322 ligated EGFP would be transformed now as that would be the first construct to get expressed anaerobically. This would mean 5 transformations would be taking place, the four pET28a ligated constructs with the highest concentrations and an EGFP ligated into pBR322.) To perform a transformation the following steps were performed:
- From each of the chosen samples first a 1ng/µL solution had to be made by diluting a portion of the sample with sterile water.
- 1µL of each of the 1ng/µL dilutions would be pipetted into 20µL of BL21 bacteria which were left on ice for 5 minutes.
- The bacteria were then heat shocked by placing them in a water bath at 42°C for 30 seconds before returning them to ice for 2 minutes.
- Hereafter, 80µL of SOC solution was added to each bacterial sample. The bacteria were now not returned to ice, rather they were incubated at 37°C for 60 minutes.
Plating
The samples which had just be transformed and incubated could now be plated onto agar plates. All would be plated onto kanamycin agar plates apart from the pBR322 ligated EGFP which would be plated onto an ampicillin plate. The plating procedure is given below:
- In the vicinity of a blue flame pipet the entire bacterial solution onto the centre of the plate.
- Using a sterile spreader spread the bacteria out over the plate.
- Incubate the plates in 37°C overnight.
This concludes the work on the four newest constructs for today.
Expression Samples
On August 19th four culture tubes had been placed in an incubator to grow overnight. Two contained pET28a ligated 1PJN and two contained pET28a ligated 1ETF. One of each these samples would be used for a control experiment when attempting to perform CEST experiments later on. The other two would now be brought across to the 2 large LB cultures (400mL) which had also been prepared on August 19th.
Controls
The two control cultures were spun down at 3700rpm for 10 minutes forming a pellet of bacteria cells. The supernatant was poured off and the pellets were placed in a +4°C refrigerator for storage.
Expression Samples
The two remaining cultures would now be used to express the proteins 1PJN and 1ETF in the larger culture mediums in the hope of obtaining a larger yield. This was attempted in the following manner:
- To start with 400µL of kanamycin was added to each of the 400mL culture mediums.
- Hereafter, the entire 10mL culture tube could be poured into one of the culture flasks.
- Now the culture mediums would be incubated at 37°C to ensure cell growth. Once there were enough cells IPTG could be added so that the bacteria would start producing our proteins. To follow the cell growth optical density measurements were performed:
| Time | Absorbance 1PJN | Absorbance 1ETF |
|---|---|---|
| 15:30 | 0.032 | 0.017 |
| 16:45 | 0.155 | 0.071 |
| 17.25 | 0.389 | 0.156 |
| 17.50 | 0.678 | 0.320 |
| 18.15 | - | 0.540 |
- Once the optical density reached a value of around 0.6 200µL of IPTG was added (for 1PJN at 17:50, for 1ETF at 18:15). This would ensure protein expression within our bacteria. The culture mediums were then kept left in the 37°C incubator overnight.
Buffer Preparation
To ensure that the protein purification could be performed in a fluid manner we decided to prepare a number some buffers for the Nickel-Column purification. We prepared three buffers: a Wash, Strip and Elution buffer using the following amounts:
| Wash Buffer | Elution Buffer | Strip Buffer | |
|---|---|---|---|
| Trizma Base | 1.21 (g) | 0.484 (g) | 1.21 (g) |
| NaCl | 14.6 (g) | 5.84 (g) | 14.6 (g) |
| Imidazole | 1.02 (g) | 6.81 (g) | - |
| EDTA | - | - | 18.6 (g) |
| Demineralised Water | 500 (mL) | 200 (mL) | 500 (mL) |
Protein Purification
After having prepared two expression mediums on August 20th it was now time to retain the proteins from the cultures and perform a purification. To do this a lot of separate steps were performed which will each be handled below in chronological order:
Protein Retention
To retain the protein from the culture the BugBuster protocol was used which was doen as follows:
- Firstly the cultures were poured into separate 400mL flasks which would fit into a large centrifuge. The flasks were equally weighed to ensure that the centrifuge was balanced.
- The flasks with the culture medium were then spun down in the centrifuge at 10000G for 10 minutes forming a pellet containing our bacteria with our protein inside. The supernatant was therefore discarded so we were left with our pellet.
Preparation for CEST measurements
To be able to measure the CEST effects of our proteins we would have to bring them to the MRI scanner. Here they would be tested on their contrast. As our bacteria would house our proteins should they be used to find tumours this is what we wished to replicate in the scanner. This would mean taking a piece of our pellet and storing it separately. The following steps therefore were taken:
- Firstly from the two large pellets we had just obtained a small portion was placed in a PCR tube and spun down at 13400 rpm for 3 minutes to place the pellet at the bottom of the PCR tube.
- We had also prepared two controls on August 20th. These were spun down to form pellets but having been left at 4°C they had dissolved again. However this solution was pipetted into a PCR tube and spun down forming a new pellet. The supernatant was then pipetted off again.
- To all four samples 250µL of 4% PFA (Paraformaldahyde) solution was added to fixate the cells pausing necrosis.
- The four samples were then allowed to sit in the refrigerator at 4°C.
Return to Protein Retention
Continuing with the remainder of the large pellet and the BugBuster protocol the following steps were then performed on the two pellets:
- To each pellet 8mL of BugBuster was added along with 8µL Benzonaze. The pellets and solution were then placed on a shaking table in 4°C for 30 minutes untill the pellet had completely dissolved.
- The solutions were then poured into a smaller flasks, weighed equally and returned to the heavy duty centrifuge where they were spun for 20 minutes at 16000G.
- The supernatant now contained our protein (along with other native proteins) so it was poured off and stored separately. The pellet was kept for analysis later on in the 4°C refrigerator.
Nickel-Column Purification
Now that we had obtained a solution containing our protein (hopefully) we could purify it using a Nickel-Column. Firstly two columns had to be prepared. To do this the following was done:
- The columns were filled with demineralised water and 1mL of resin was added to each column.
- The water was allowed to flow through but was refilled before the column was emptied. As the resin layer grew more resin was added to the column.
- The above procedures were repeated until a 2mL thick resin layer had formed in the column.
- Now a filter could be placed on top of the resin using a plunger.
Having prepared a column it was time for the purification to begin:
- First the column was loaded with 8mL of Charge buffer containing 50mM NiSO4. The flow through of the column was stored separately due to the heavy metal content.
- Once empty 16mL of Wash buffer was loaded and again the flow through was separately caught to reduce the risk of heavy metal contamination.
- The entire protein sample could then be loaded onto the column. Of the flow through a single droplet was retrieved for future analysis.
- The column was then washed once more with 16mL of wash buffer and again a single droplet was caught for analysis.
- Now elution was performed by loading the column with 8mL of elution buffer. All the flow through droplets were retrieved by catching 0.5mL of flow through in separate eppendorf tubes.
- The column was then stripped with 8mL of strip buffer, whereby the flowthrough was retrieved separately due to the Nickel content. Strip buffer was then added to the column before sealing it and placing it in the 4°C refrigerator.
Our purified protein could now be found in the 16 elution tubes we had collected (in reality we had collected 14 tubes from the 1PJN elution and 16 tubes from the 1ETF solution).
Concentration Determination
Now we had our protein it was important to know what the concentration was. As we could not measure the 260/280 absorbance, a nanodrop measurement was not applicable. We had to think of a way to determine the concentration of heavily positive proteins which led us to the Bradford assay. The following steps were performed to achieve this:
Making the Protein Reagent
For the staining of our samples we made a solution consisting of Coomassie Brilliant Blue G-250, 95% Ethanol and 85% Phosphoric Acid.
- First 20mg of Coomassie Brilliant Blue G-250 was weighed and dissolved in 10ml of 95% (w/v) Ethanol.
- Next we added 20ml of (85% (w/v) Phosphoric Acid and diluted the solution until a total volume of 200ml was reached.
- The solution was subsequently stirred for about an hour, to ensure that the Coomassie Brilliant Blue G-250 was completely dissolved.
Making the BSA standards
To ensure we had reference dilutions to work with the following BSA (Bovine Serum Albumine) solutions were prepared. These would allow us to compare our protein concentrations with those of the standards, giving an indication of the concentration of our protein.
| Samle # | Standard Concentration (mg/mL) | Standard Volume (µL) | Dilent Volume (µL) | Final Protein Concentration (µg/mL) |
|---|---|---|---|---|
| 1 | - | - | 500 | 0 |
| 2 | 2 | 0.67 | 499.33 | 1.25 |
| 3 | 2 | 1.25 | 498.75 | 2.5 |
| 4 | 2 | 2.5 | 497.5 | 5.0 |
| 5 | 2 | 3.33 | 496.67 | 10.0 |
| 6 | 2 | 3.75 | 496.25 | 15.0 |
| 7 | 2 | 5.0 | 495.0 | 20.0 |
| 8 | 2 | 10 | 790.0 | 25.0 |
In the above table the standard refers to the BSA solution and the Dilent used was a pH 8.0 TRIS buffer.
Performing the Bradford Assay
To perform the Bradford assay a 96 wells plate was used and filled in the following manner:
- 100µL of each of the above standards was pipetted into one of the wells on a 96-well-plate.
- To each standard 100µL of the Coomaissie dye was added for colouring.
- We then loaded 50µL from each of the Nickel column elution flow through tubes. (We discarded the first 2 and last 2 samples from each protein as these would not accurately represent our proteins).
- To this 50µL we then added 50µL of TRIS buffer (pH 8.0).
- As with the standards 100µL of the Coomassie Dye was then added.
These dilutions were not very clear so it was decided to perform the test again but this time taking only 10µL from each of the samples and add 90µL of TRIS buffer. This allowed us to see clearly which of the sample tubes contained the highest concentration of our two proteins: 1PJN and 1ETF.
The wells plate was then not placed in the spectrometer as we could clearly see which of the collection columns contained the highest protein concentration from each of the protein elutions. The five highest concentrations were then chosen for the next steps. The 96-Wells-Plate used for the assay is shown below:
Rebuffering the Proteins
As the proteins had been purified using a Nickel Column they were now suspended in a buffer containing Imidazole which can cause protein denaturation. This meant that the proteins had to be rebuffered into a more suitable buffer:
- First, from the five chosen elution samples 10µL was pipetted into a separate eppendorf tube for an SDS analysis later on.
- Then the five chosen elution samples from each protein were combined in one large falcon tube.
- The rebuffering was then performed using a PD-10 desalting column with the following protocol:
- First the column was opened and the buffer was poured off and discarded.
- Next 25mL of TRIS buffer (pH 8.0) was allowed to run through the column. The flow through was discarded.
- After this we were able to load our sample, during which the flow through was discarded. A maximum of 2.5mL of our sample could be loaded, which was the exact amount we had chosen to rebuffer.
- Then 3.5mL of TRIS Buffer was run through the column for the elution, and the flow through was retained as it contained our protein. Retention was done in a number of eppendorf tubes each holding 1mL of the flow through.
- The proteins were then snap frozen in liquid nitrogen and stored in -80 degrees Celsius.
SDS Analysis
Today we would analyse the results of the protein purification performed on August 21st. This would be done by running an SDS-PAGE gel. As we were not certain if the super positive proteins would like the conditions of a standard SDS PAGE gel we decided to also test our samples on a native gel. To further enhance this we would attempt to run this gel with the poles reversed, in theory forcing the protein down the gel.
- To start with two 12% gels were prepared. The SDS PAGE gel followed the standard recipe whereas the native gel would not contain the 10% SDS in either the running or stacking gel solution.
- Once the gels had hardened the samples could be prepared. We would like to load a sample from the loading, washing, the five chosen elution samples and a sample from the original pellet which we had also stored on August 20th.
- For both proteins, for each of the 5 elution samples and for the loading and washing samples 2 PCR tubes were filled with 5µL of each sample.
- To 1 of each pair of PCR tubes (containing the same sample) 5µL of standard 2x SDS loading dye with DTT (stock: 500µL 2x SDS loading dye, 1µL DTT) was added.
- To the other PCR tube of the pair 5µL of SDS free loading dye was added.
- Then a single eppendorf tube per protein was loaded with a small pellet scraping and 100µL of pH 8.0 TRIS buffer was added.
- Of the pellet solution 2x 5µL was then pipetted in to separate PCR tubes and 5µL of the correct loading dye was added to each of the four tubes.
- Now all the samples containing the SDS loading dye were denatured for 5 minutes at 95°C.
- Now the samples had been prepared the gels were placed in the tanks and surrounded with buffer. For the SDS gel 1x SDS buffer was added. For the SDS free gel an SDS free buffer was used.
- The gels were now run for 15 minutes at 90V and then for 40 minutes at 150V.
- After the gels had run they were placed in water and allowed to sit for 30 minutes before being stained with 1x Coomaisse solution for two hours.
- The gels were then set in demineralised water and left on the shaking table overnight.
Preparation for Anaerobic Expression
It was finally time to work on the anaerobic expression. Over the past few weeks a lot of effort had gone into fixing up an old biofermentor making it usable once more. Now we had it up and running we were going to test it by attempting to express EGFP anaerobically. Firstly however a large culture solution had to be prepared and small cultures had to be grown for expression to be kick started.
Preparing Culture mediums
In the biofermentor a 4L LB solution was prepared before the fermentor was autoclaved. This solution was prepared in the vessel by mixing:
- 40g Peptones
- 40g NaCl
- 20g Yeast Extract
- 4L Demineralised Water
The fermentor was then autoclaved. Along with it a smaller 400mL culture medium was autoclaved. This could be used to grow EGFP aerobically to check that no proteins would grow if oxygen was present. For this 400mL solution the following were mixed:
- 4g Peptones
- 4g NaCl
- 2g Yeast Extract
- 400mL Demineralised Water
Preparing Small Cultures
To ensure that the bacteria did not have to grow from scratch in the two larger culture mediums smaller culture tubes were prepared. To do this we retrieved our pBR322 ligated EGFP agar plate from the refrigerator and followed the following protocol:
- Two eppendorf tubes were filled with 15µL of sterile water.
- The following steps were all performed in the vicinity of a blue flame.
- 2 colonies were picked from the above mentioned agar plate and each was dissolved in one of the water samples.
- Then 7 culture tubes were filled with 10mL of LB medium.
- To each culture tube a further 10µL of Ampicillin antibiotics was added.
- Then to 6 of the 7 tubes 5µL of the dissolved picking solutions was added.
- To the 7th culture tube a single picking was taken and was ejected directly, including the pipet point, into the culture medium.
- The culture tubes were then placed in the 37°C incubator and left to grow overnight, in preparation for tomorrows expression.
Six of the above prepared culture tubes would be used for the 4L anaerobic expression and the final tube would be used for the control.
MRI Experiments
On August 21st we had prepared a number of samples for testing in the MRI. These four pellets (3 1ETF and 1 1PJN) were stored in 4% PFA solution and left in the 4°C refrigerator overnight. Now they were transferred to the MRI laboratory for analysis of their CEST effect. Of the three 1ETF samples 1 had had protein expressed and 2 were controls without protein expression. The 1PJN sample had also endured protein expression. Of the two controls one had had 100µL of 0.01% Poly(Lysine) added to spike the sample. The MRI experiments were then performed using the following protocol:
- The four samples were placed and secured in the MRI and a standard scan was performed.
- The frequencies were then optimized for our proteins.
- We then shimmed the samples, which means we created a homogeneous magnetic field.
- The T2 was weighed for the orientation.
- We then performed local shimming on each of the separate pellets.
- We then averaged our measurements over 8 separate scans, without inducing a saturation pulse.
Using these measurements large amounts of data were acquired which the modelling teams would analyse, giving us the results of our proteins.
Protein Expression
Today we would focus on the anaerobic expression of the pBR322 ligated EGFP. This should give the first indication of whether our promoter works or not. On August 22nd we had autoclaved two culture mediums, a 4L medium in the biofermentor to attempt anaerobic expression and a 400mL medium for an aerobic control experiment. Before beginning 4mL and 400µL of Ampicillin was added respectively to each of the above mentioned cultures. Before beginning with our experiments we had to calibrate the oxygen probe which was done in the following manner:
- Calibration at 100% by flooding culture vessel with air for 30 minutes (3L/minute, 300rpm agitation).
- When gas admission was increased to 10L/minute the dO2 increased from 100% to 107.9%.
- Calibration was repeated at 10L/minute air admission.
- Once gas admission was removed the dO2 stabilized at 92.0%. This value was then taken as the 100% calibration.
- For the 0% calibration the Oxygen probe was disconnected.
Now we could begin with the protein expression. Multiple steps and measurements were taken at multiple time points. Brief descriptions of the steps have been given in the following table:
| Time | Procedure |
|---|---|
| 09:02 | Single small culture tube prepared on August 22nd was added to the culture medium and medium was placed in 37°C aerobic incubator. |
| 09:16 | First of 6 small culture tubes added to 4L anaerobic culture chamber. (pH = 7.78, dO2 = 99.5, Temp = 36.7°C) |
| 09:28 | Last of 6 small culture tubes added to the 4L anaerobic culture chamber. |
| 10:03 | Aerobic culture sampled. O.D. = 0.080. |
| 10:11 | The dO2 of the anaerobic chamber had fallen to 53.8. Therefore 100% air was implemented into the chamber at 3L/minute. |
| 10:24 | Following equilibrium had occured (pH = 7.75, dO2 = 78.8, Temp = 36.9°C). |
| 10:27 | Anaerobic culture was sampled. O.D. = 0.084. |
| 10:37 | Anaerobic chamber conditions = (pH = 7.72, dO2 = 71.6, Temp = 37.0°C) |
| 10:50 | Anaerobic chamber conditions = (pH = 7.67, dO2 = 60.6, Temp = 37.0°C). The low Oxygen saturation meant we upped the gas induction to 5L/minute. |
| 11.35 | Anaerobic chamber conditions = (pH = 7.45, dO2 = 36.3, Temp = 37.0°C). Low Oxygen saturation forced a gas induction of 10L/minute. |
| 11:42 | Anaerobic chamber conditions = (pH = 7.44, dO2 = 73.7, Temp = 36.9°C). Higher Oxygen saturation resulted in a reduction of gas induction to 8L/minute. |
| 11:48 | Anaerobic culture was sampled. O.D. = 0.0511 |
| 11.53 | Aerobic culture was sampled. O.D. = 1.087 |
| 11:58 | Anaerobic chamber conditions = (pH = 7.25, dO2 = 16.1, Temp = 37.0°C). |
| 12.04 | Anaerobic chamber conditions = (pH = 7.14, dO2 = 30.3, Temp = 37.0°C). Agitation was reduced to 250rpm. |
| 12:15 | pH loop was induced. (Setpoint = 7.5, Deadband = 0.2, Multiplier = 50%) |
| 12:15 | Anaerobic chamber conditions = (pH = 6.97, dO2 = 45.4, Temp = 37.1°C). |
| 12:18 | Anaerobic chamber conditions = (pH = 7.18, dO2 = 57.4, Temp = 37.0°C). |
| 12:22 | dO2 = 40.2. As the optical density was now presumable around 0.6 we could begin reducing the oxygen levels, hopefully inducing protein expression. (Gas = 4Gas) |
| 12:25 | Anaerobic chamber conditions = (pH = 7.84, dO2 = 0.2, Temp = 37.1°C). |
| 12:25 | Sample of anaerobic chamber was taken (λ = 1). |
| 12:31 | Anaerobic chamber conditions = (pH = 7.86, dO2 = -0.7, Temp = 37.0°C). The 4Gas induction was set to 5L/minute. |
| 12:36 | Aerobic sample was taken. (µ = 1). |
| 12:42 | Anaerobic chamber conditions = (pH = 7.90, dO2 = -0.4, Temp = 36.9°C). 0.1M H2SO4 was connected to the acid inducer. pH multiplier was set to 20%. |
| 13:00 | Anaerobic chamber conditions = (pH = 8.02, dO2 = 0.2, Temp = 37.0°C). |
| 13:02 | Anaerobic sample was taken. (λ = 2). |
| 13:14 | Anaerobic chamber conditions = (pH = 7.56, dO2 = 1.2, Temp = 37.0°C). |
| 15:16 | Anaerobic chamber conditions = (pH = 7.45, dO2 = -0.7, Temp = 37.0°C). |
| 15:22 | Anaerobic sample was taken. (λ = 3). |
| 15:25 | 4Gas was switched to 100% Nitrogen gas. Agitation was set to 100rpm to reduce foaming. |
| 17:03 | Anaerobic sample was taken. (λ = 4). |
| 18:05 | Anaerobic sample was taken. (λ = 5). |
| 18.07 | Heater was set to 20°C. |
| 18:24 | Gas admission was turned off. pH control was also turned off, pH = 7.64. |
| 18:38 | Temperature was 25.5°C. |
After the fermentor had been turned off all samples we poured into specialised centrifuge tubes and equally weighed. These were then centrifuged for 10 minutes at 16000g. The samples were then snap frozen and placed in the -30°C freezer. and left for later analysis.
Week 6
Expression Cultures