 "
"
Team:Paris Bettencourt/Project/Sabotage
From 2013.igem.org








Background
One of the main concerns about tuberculosis today is the emergence of antibiotic resistant strain.
Results
- Construction and characterization of phagemids coding for small RNA targeting antibiotic resistance proteins.
- Showed theoretically burden of a device is critical for the maintenance of a genetic element in a population.
- Successful conversion of antibiotic resistant population of E. coli to a sensitive state.
BioBricks
Aims
Our objective is to make an antibiotic-resistant bacterial population sensitive again to the same antibiotic.
Skip to Design
Skip to Results
Skip to Perspectives
Introduction
As antibiotic-resistant bacterial strains are rising and pharmaceutical pipelines are drying up, we decided to invest in a new strategy based on specific silencing of the genes responsible for resistance through bio-engineered stealth bacteriophages. The silencing of the genes is obtained with a simple and modular system of tailor-made small RNAs. The spreading of this construct in a bacterial population is based on an autonomous phagemid/helper phage system. We demonstrated the validity of this trojan horse strategy by converting back to a sensitive state populations of bacteria initially resistant to antibiotics as chloramphenicol or kanamycin.
 Figure 1. The Trojan Horse Strategy : Infecting the cells with a phage bearing a sRNA silencing the expression of antibiotic resistant genes converting it back to a sensitive state.
Figure 1. The Trojan Horse Strategy : Infecting the cells with a phage bearing a sRNA silencing the expression of antibiotic resistant genes converting it back to a sensitive state.
Design
Designing a silencing device
Most antibiotic-resistance result from specific bacterial proteins such as efflux pump that extract the antibiotics from the cells or enzymes that quickly metabolize the antibiotic molecules. We choose a straightforward strategy to force bacteria to stop producing those proteins by targeting their cognate mRNA with tailor-made small RNAs. In order to design our silencing device, we used the protocol described in Na et al (2013) and inserted a 24bp sequence complementary to the RBS area of the target genes inside the published scaffold sequence that allow the stabilization of the hybridization through recruitment of the Hfq protein. We designed three sRNA respectively targeting Kanamycin resistance gene located on a pCOLA Duet Vector, Chloramphenicol resistance on pACYC Duet Vector and lacZ on E. coli's chromosome.

Figure 2. Structure of the Chloramphenicol resistance silencing module
Designing a genetic element that spread in a population
We adopted non-lytic filamentous bacteriophages as our vectors of choice as they are good at spreading genetic elements in bacterial populations. However, being infected by a phage represents a huge burden for an individual bacteria which we thought would be detrimental for our construct to be able to maintain itself long enough in a population for this system to be clinically relevant.
Therefore we chose a phagemid/helper system which is composed of two mobile interacting elements. The “helper” elements of this system is an M13 phage mutant that provides packaging proteins but largely lacks the capacity to be packaged; the “light” element called phagemid is a normal plasmid harbouring the packaging signal and can thus propagate from hosts carrying the “helper” phage.
As a result a cell containing both elements will produce and secrete many encapsulated phagemids together with a small minority of helper phages.
We expect such a system to infect with phagemids the majority of cells in a population and to be able to spread and maintain itself thanks to a small number of co-infected cells harboring helper phages, transforming them into phage-producing cells.
Our silencing device is cloned on the light phagemid element in order to spread it efficiently. As the post-transcriptional regulation we are using only rely on RNA, it does not require protein synthesis, resulting in a very low cost for the cell. Moreover producing protective proteins against antibiotics is costly for the cell and lowers its fitness in an antibiotic free environment, we thus expect our silencing device to be a temporary relief for the infected cell which should avoid early counter-selection dynamics.
Design of a sequential killing strategy
To perform our proof of concept experiments, our targeted genes were different antibiotic resistance genes on commercial plasmids that were transformed in E. coli MG1655 cells. Just as in nature where resistance genes are often found on mobile elements, our model respects the constraint of having multiple copies of the target gene to silence which would not have been the case with a chromosome located target.
The experimental set up consisted of a sequential protocol: (I) Phage infection leads to the silencing of the antibiotic-resistance genes and (II) cells are plated on antibiotics to kill the sensitive phenotype and quantify the efficiency of the silencing.
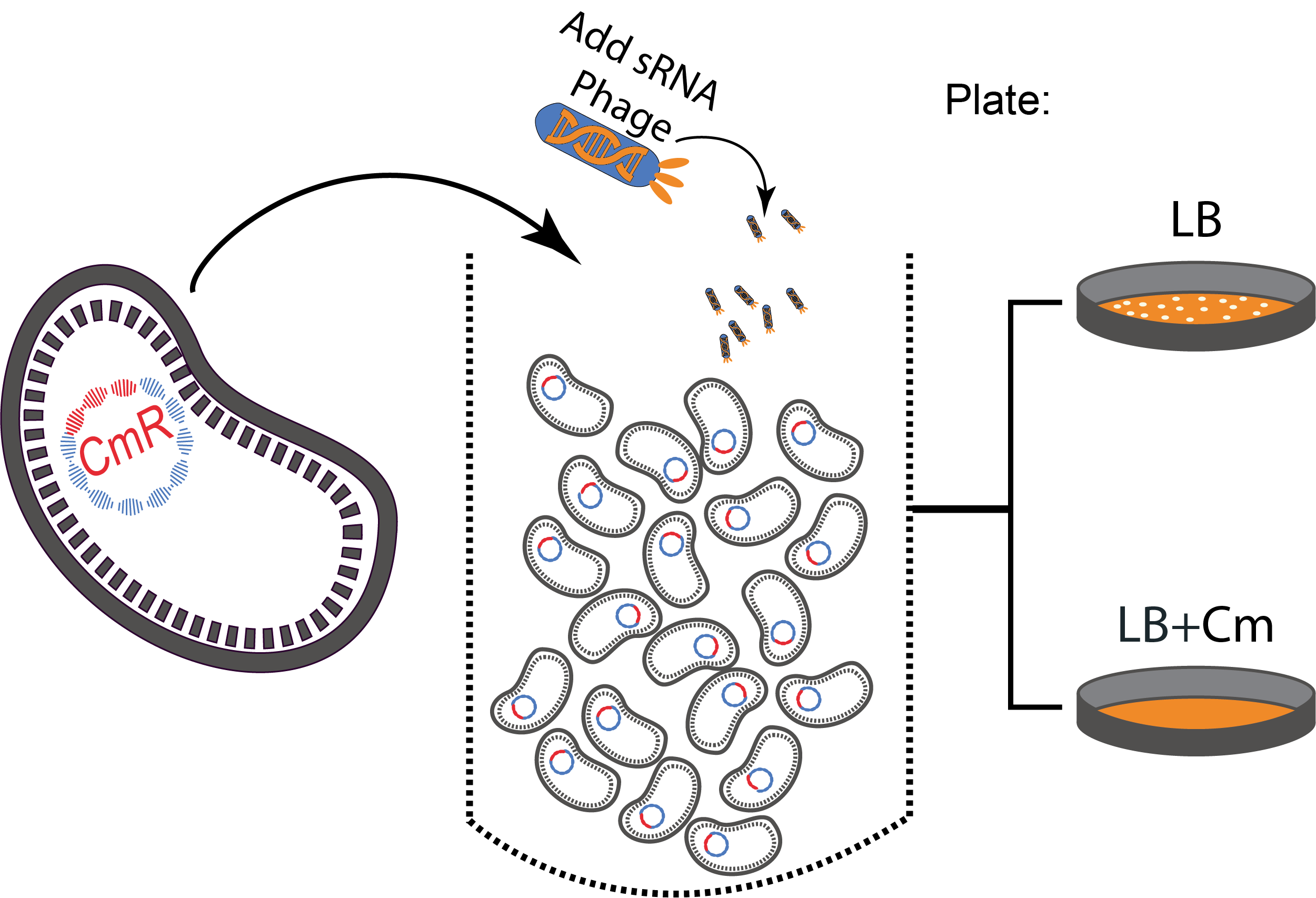 Figure 3. A sequential killing strategy :
Figure 3. A sequential killing strategy : Characterization of the phagemid system
The phagemid Helper system (kindly provided by Monica Ortiz of D. Endy lab) consists of a helper plasmid (M13K07) bearing Kanamycin resistance and a phagemid (Litmus-28) bearing GFP and an Ampicilin resistance cassette. Using those different markers, we characterized the system by infecting the cells with a ratio of phage of 1 to 100 and plating appropriate dilutions on different medias (LB, Kan, Amp, Kan/Amp). We show that the phagemid is spread a thousand fold more than the helper plasmid. There are as many cells infected with the helper as cells infected by both helper and phagemid, that is to say, phages producing cells. Those results show the validity of using this system to spread a genetic element in a bacterial population. Most of the cells bear the desired genetic element and the spread is carried on by a minor quantity of cells producing the vector.
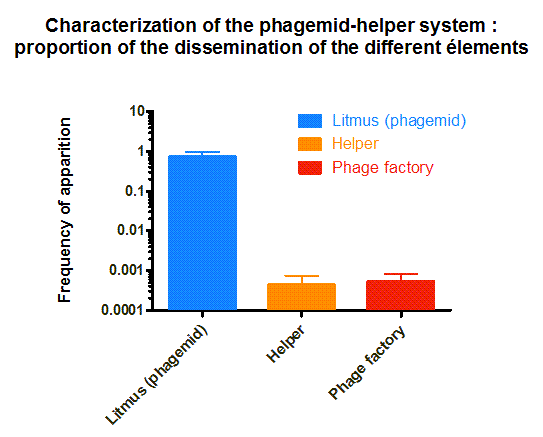 Figure 4. Characterization of the phagemid helper system
Figure 4. Characterization of the phagemid helper system
Effect of device burden
Every introduced system to a cell will consume resources and compete for internal molecular machinery. In case the device provides no direct benefit for the organism, the burden of the device will lead to a decrease in growth rate, and therefore a lower fitness. One way to avoid out-competition due to lower growth rate is by horizontal gene-transfer. We studied the behavior of a the phagemid/helpel system, which is a derived from from the non-lytic bacteriophage M13. This systems propagate through E. coli F+ populations via two vectors: a phagemid carrying the desired device, and a helper carrying the necessary machinery for production of the bacteriophage. Only when both vectors are present in a cell it will produce bacteriophage (see diagram on the right). For more details please go to here.
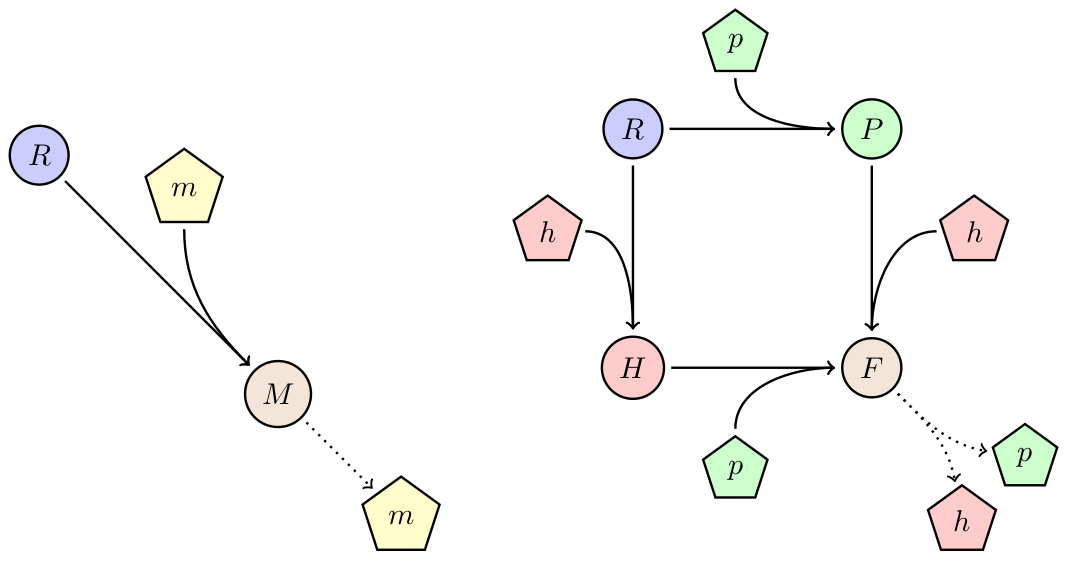
Figure 5:
We found that our system should be stable in time, even under chemostat conditions. At common parameters found in the literature, we found that the burden of the device has no strong effect. However, relatively low variations (one order of magnitude reduction), makes small variations in growth rate extremely relevant for the time the infected populations remain in the system. We expect that the phage production rates under limiting conditions such as the lungs to be smaller than those described in liquid cultures, making desirable to reduce growth rate as much as possible.
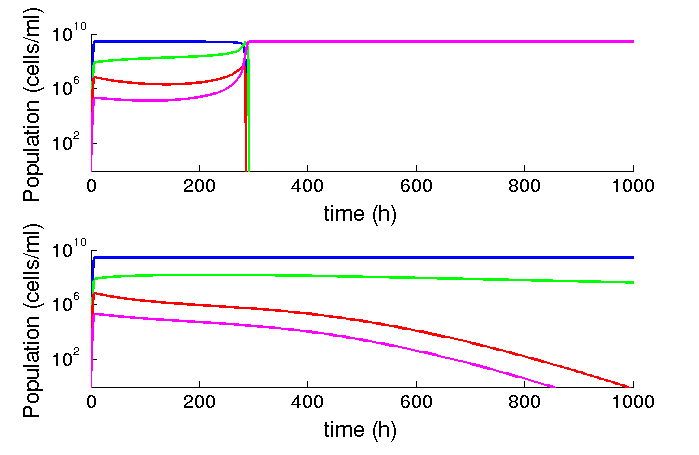
Figure 6: Al lower phage production rates, relatively low differences in burden matter
Making a Chloramphenicol resistant E. Coli population sensitive to Chloramphenicol
We used our designed phagemid, bearing the sRNA anti-Cm, to infect a population of chloramphenicol resistant cells. By applying the sequential killing strategy described in Figure 3, we efficiently killed 99,1% of the bacterial population (chloramphenicol concentration 1000ug/ml). We tested this strategy with different concentrations of chloramphenicol. We show that the rise of chloramphenicol concentration directly lead to drastic reduction of the survival of the infected cells.
In order to show the general application of our system, we then applied the same strategy on MG1665 transformed with pCOLA Duet. Those kanamycin resistant cells were infected with phages bearing our anti-Kan sRNA. 99,87% of the population was killed (standard deviation 0,23 %) at 1000ug/mL of kanamycine

Figure 7. Killing Chloramphenicol resistant and Kanamycin resistant E. Coli population with antibiotic
Charaterization of the origin of resistance
One of the main limits of our system could be the development of resistances. Indeed, there are at least two ways to become resistant to our system: by avoiding infection or by developing a resistance to the silencing. In order to get a better idea of what is the main cause of resistance we screened the survivor colonies for GFP expression. GFP positive colonies correspond to infected cells with malfunctioning silencing. We show that 70% of resistance is caused by a resistance towards the sRNA. This resistance could be due to a mismatch between the sRNA sequence and the target sequence resulting from a single point mutation.

Figure 8. Origin of resistance to the sequential killing
Conclusion and perspectives
Several sRNA on the same phagemid : Although our silencing small RNAs were assembled in phagemids with Gibson assembly, the Biobricks cutting sites included in our BBG adapter method provide full modularity. In our case a direct application is the assembly of multiple silencing modules (promotor+sRNA+terminator) on a single phagemid vector. As the main clinical problem with bacteria is multi or even pan-resistance this modularity provide an interesting strategy to explore as it allows simultaneous downregulation of multiple target genes.
Building a library of sRNA : As our silencing strategy rely on base pair complementarity, a main concern from the clinical point of view would be the fast emergence of resistances to silencing through single point mutations. Indeed even if the majority of mutations that would decrease the efficiency of the silencing would also lead to an impaired or non functionnal protective protein, some neutral mutations will still be made possible by the redundancy of the genetic code. To tackle this issue an interesting solution would be to build a library of small RNA containing single point mutation on those particular neutral positions and load it on our phagemids. By being one move ahead of evolution we expect that such an anticipating strategy would considerably slow down the emergence of resistance to silencing.
Possible applications : As most bacteria species have their own phages, our strategy could be adapted to a broad range of diseases. Tuberculosis of course is particularly concerned because the number of panresistance cases is exploding in hospitals but potential targets could also be Staphylococcus aureus infections or even dangerous strains of E. coli as suggested in our model.
sRNAs as bioricks, an adaptable silencing tools : We biobricked the sRNAs described in that study with their scaffold. Since the targeting sequence of the system is only 24 bp, it is very easy to engineer new sRNA by using a classic mutagenic PCR technique (Seung Min et al, 2013). This means that every iGEM team is now one PCR away from silencing any gene it wants .
Literature
- Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nature Biotechnology(2013).
- Monica E Ortiz and Drew Endy. Engineered cell-cell communication via DNA messaging. Journal of Biological Engineering (2012).
- Seung Min Yoo, Dokyun Na and Sang Yup Lee. Design and use of synthetic regulatory small RNAs to control gene expression in Escherichia coli. Nature Protocols 8, 1694–1707 (2013).
- Woodford N, Wareham DW; UK Antibacterial Antisense Study Group. Tackling antibiotic resistance: a dose of common antisense? The Journal of Antimicrobial Chemotherapy. 2009 Feb;63(2):225-9.
Attributions
The project itself was designed and accomplished by Clovis Basier, Aude Bernheim and Vincent Libis in consultation with Edwin Wintermute and Ariel Lindner. Modeling was done by Sebastian Jaramillo-Riveri.
Most of the strains (MG1655, MG, MGZ1, NED turbo) and plasmids (Duet vectors, pUC18) used for this project were kindly provided by the INSERM U1001 lab.
The phagemid template as well as the helper plasmid were provided by Monica Ortiz from the Endy Lab, Stanford.
We want to thank each member of the team Paris Bettencourt who worked with us and permit this project !

 +33 1 44 41 25 22/25
+33 1 44 41 25 22/25
 team2013@igem-paris.org
team2013@igem-paris.org 
