 "
"
Team:TU-Eindhoven/CESTAgentTesting
From 2013.igem.org
(→Cloning) |
(→Cloning) |
||
| Line 24: | Line 24: | ||
{{:Team:TU-Eindhoven/Template:ImageList}} | {{:Team:TU-Eindhoven/Template:ImageList}} | ||
| - | {{:Team:TU-Eindhoven/Template:Float | position=left | size= | + | {{:Team:TU-Eindhoven/Template:Float | position=left | size=8 }} |
{{:Team:TU-Eindhoven/Template:Image | filename=colpcrpet28a.jpg}} | {{:Team:TU-Eindhoven/Template:Image | filename=colpcrpet28a.jpg}} | ||
{{:Team:TU-Eindhoven/Template:FloatEnd | caption=Colony PCR of the ligated constructs into pET28a| id=colpcrpet }} | {{:Team:TU-Eindhoven/Template:FloatEnd | caption=Colony PCR of the ligated constructs into pET28a| id=colpcrpet }} | ||
Revision as of 12:37, 4 October 2013


Contents |
Abstract
Introduction
Initially, a total of 10 constructs with proteins were designed, all containing a high percentage of lysine and/or arginine, which have a chemical shift which can be used for CEST imaging. Four of them were naturally occurring proteins, native to either E. coli or to other organisms, in which case the protein sequence was optimized to match codon prevalence in E. coli. These were 1G70, 1ETF, 1PJN Chain-1 and Human Protamine 1 (HPM1, optimized). The other five were non-natural polypeptides consisting of a two repeating dipeptides, namely poly(arginine-glycine) (P(RG)), poly(lysine-serine) (P(KS)), poly(arginine-serine) (P(RS))and poly(threonine-lysine) (P(TK)). Like HPM1, these we E. coli optimised. Additionally, an EGFP construct would also be inserted and expressed as a positive control.
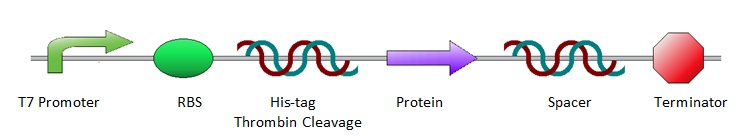
All the protein sequences were cut out of the ordered pUC57(s) vectors by NheI and XhoI restriction enzymes and ligated into pET28a vectors. As a result, all constructs shared the same lay-out, starting with prescribed iGEM restriction sites EcoRI, NotI and XbaI, followed by an T7 promotor site and ribosome binding site. Hereafter are a poly-histidine tag (for protein purification), thrombin cleavage site and NheI restriction site (to isolate the protein sequence for ligation into a pET28a vector). Subsequently comes the variable part, the protein coding sequence. Then follow an XhoI restriction site and random spacer sequence, positioning the terminator at 30 base pairs from the stop codon. After the terminator come three prescribed iGEM end restriction sites, SpeI, NotI, PstI, plus a HindIII restriction site.
Methods and Results
Cloning
Once the pUC57(s) constructs were delivered, we could transform them into NB bacteria for amplification of the vectors. When they were amplified, a digestion on the constructs as well as on the pET28a vector is performed with NheI and XhoI restriction enzymes to cut out just the protein sequence out of the pUC57(s) construct. Chosen was for pET28a since it has a promoter that can be induced by IPTG which is helpful and on the otherhand there was much DNA left in stock. Once the pUC57(s) constructs as well as pET28a vector were digested a gel extraction was performed to cut out the desired bands. Then a ligation is performed to get the protein sequence from the pUC57(s) constructs into the pET28a vector. After ligation the constructs were plated with kanamycin antibiotics and grown overnight to create colonies. To check whether ligation was succeeded a colony PCR was performed. Primers used in the ligation of pET28a vectors were the T7 Forward and T7 Reverse (terminator) primers.
After completing the PCR protocol, an agarose gel electrophoresis was executed on the products and visualised using UV spectrometry to see whether the ligation was successful or not.
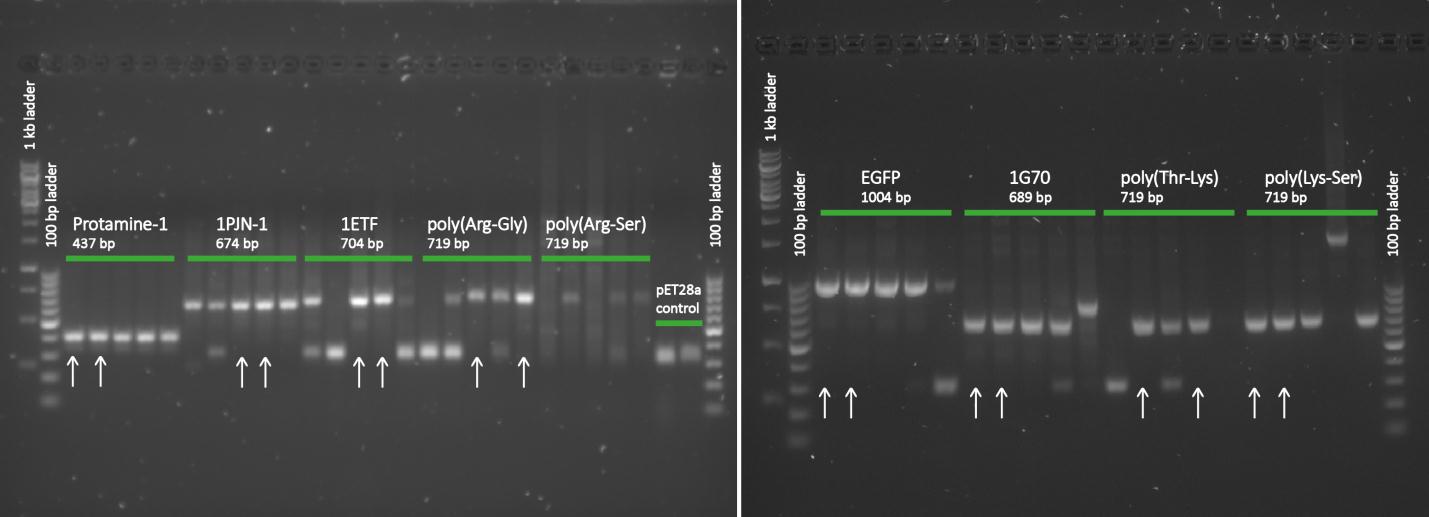
Except for the poly(arginine-serine), all constructs yielded successfully ligated products for further processing. For each protein sequence, two of these were selected based on the intensity and the accurate weights of the appearing bands. Then from the chosen constructs, small cultures were set in, again with kanamycin, and grown overnight. To retain the vectors, a miniprep procedure was performed and once the constructs were sequenced and seemed to be correct, they were transformed into BL21 bacteria for protein expression.
Protein Expression and Sampling for CEST Measurements
Large cultures with the BL21 bacteria, containing our constructs, were set in and were grown until the optical density was about 0.6. Then protein expression was induced by adding IPTG to induce the promoter and protein expression was then performed overnight at 37 °C.
For protein retention a Bugbuster protocol was performed. Once pellets had formed during the Bugbuster protocol, samples of the pellets were taken for CEST measurements. This was done by taking about 300-400 mg of the pellet (this was done to make comparison of the CEST measurements possible) and the pellets were placed into a PCR tube and the cells were fixated with 4% PFA (paraformaldehyde) and placed in the refrigerator at 4°C for at least two days.
After the Bugbuster protocol a Nickel-column purification was performed to purify the proteins (since they have an His-tag). Samples were taken from each step (load, wash and elution) for further analysis (SDS gel analysis and Bradford assay), after rebuffering to TRIS buffer, since imidazole can cause protein denaturation.
Since it was not certain if the SDS gel analysis would be successful (the proteins were extremely positively charged, since they were selected for that property to generate CEST contrast), it was decided to run a native gel for 1ETF and 1PJN.
MRI Measurements
Discussion and Conclusions