 "
"
Team:Evry/Protocols/03
From 2013.igem.org
m |
|||
| (24 intermediate revisions not shown) | |||
| Line 9: | Line 9: | ||
<h1> Plasmid purification </h1> | <h1> Plasmid purification </h1> | ||
| - | <h2> | + | <h2> Goal </h2> |
| - | <p>The aim of the purification step is to recover the plasmid produced by the bacteria (cloning strain).<br> | + | <p>The aim of the plasmid purification step is to recover the plasmid produced by the bacteria (cloning strain).<br> |
| - | The different step are use to throw other bacterial components off.</p> | + | The different step are use to throw other bacterial components off (proteins, cell wall, genomic DNA, etc).</p> |
<h2> Preparation </h2> | <h2> Preparation </h2> | ||
| - | <i> | + | <i>Protocol adapted from Macherey-Nagel plasmid purification notebook</i><br><br> |
| - | <b>1. Cell culture</b><br> | + | <b><p>1. Cell culture</b><br> |
| - | Cultivate cells in LB medium overnight.<br>< | + | Cultivate cells in LB medium overnight.<br></p> |
| - | <b>2. Cell harvesting</b><br> | + | <p><b>2. Cell harvesting</b><br> |
| - | Set saturated E.coli LM culture into 2 mL tubes. Centrifuge at 11 000 x g for 30 secondes. Discard as much as supernatant as possible.<br>< | + | Set saturated E.coli LM culture into 2 mL tubes. Centrifuge at 11 000 x g for 30 secondes. Discard as much as supernatant as possible.<br><p> |
| - | <b>3. Cell lysis</b> <br> | + | <p><b>3. Cell lysis</b> <br> |
| - | Add 250 μL Buffer A1 (resuspension buffer). Resuspend the cells with a vortex or a pipette. | + | Add 250 μL of Buffer A1 (resuspension buffer). Resuspend the cells with a vortex or a pipette. |
<br> | <br> | ||
| - | Add 250 μL Buffer A2 (lysis buffer). Mix gently by inverting the tube 6 - 8 times. Incubate at room temperature until lysate appears clear.<br> | + | Add 250 μL of Buffer A2 (lysis buffer). Mix gently by inverting the tube 6 - 8 times. Incubate at room temperature until lysate appears clear.<br> |
| - | Add 300 μL Buffer A3 (neutralisation buffer). Mix thoroughly by inverting the tube 6 - 8 times .<br>< | + | Add 300 μL of Buffer A3 (neutralisation buffer). Mix thoroughly by inverting the tube 6 - 8 times .<br></p> |
| - | <b>4. Lysate clarification</b><br> | + | <p><b>4. Lysate clarification</b><br> |
| - | Centrifuge at 11 000 x g for 5 minutes . Repeat this step until supernatant is not clear.<br>< | + | Centrifuge at 11 000 x g for 5 minutes . Repeat this step until supernatant is not clear.<br></p> |
| - | <b>5. DNA Binding<br></b> | + | <p><b>5. DNA Binding<br></b> |
| - | Place a NucleoSpin Plasmid Column in a Collection Tube of 2 mL | + | Place a NucleoSpin Plasmid Column in a Collection Tube of 2 mL and set the supernatant from the last step. Centrifuge at 11 000 x g for 1 minute. Discard flow-through and place the column back into the collection tube.<br></p> |
| - | <b>6. Membrane washing<br></b> | + | <p><b>6. Membrane washing<br></b> |
| - | Add 600 μL Buffer A4 (wash buffer) previously supplemented with ethanol. Centrifuge ar 11 000 x g for 1 minute. Discard flow-through and place the column back into an empty collection tube.<br>< | + | Add 600 μL of Buffer A4 (wash buffer) previously supplemented with ethanol. Centrifuge ar 11 000 x g for 1 minute. Discard flow-through and place the column back into an empty collection tube.<br></p> |
| - | <b>7. Dry membrane<br></b> | + | <p><b>7. Dry membrane<br></b> |
| - | Centrifuge at 11 000 x g for 2 minutes and discard the collection tube.<br>< | + | Centrifuge at 11 000 x g for 2 minutes and discard the collection tube.<br></p> |
| - | <b>8. DNA Elution<br></b> | + | <p><b>8. DNA Elution<br></b> |
| - | Place the column in a 1,5 mL and add 50 μL de Buffer AE (elution buffer). Incubate at room temperature and centrifuge at 11 000 x g for 1 minute.<br> | + | Place the column in a 1,5 mL and add 50 μL de Buffer AE (elution buffer). Incubate at room temperature and centrifuge at 11 000 x g for 1 minute.<br></p> |
<h2> Test </h2> | <h2> Test </h2> | ||
| - | <p>Before the storage of the tubes at -20°C, measure the concentration with nanodrop. | + | <p>Before the storage of the tubes at -20°C, measure the concentration with nanodrop.</p> |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | If the concentration is between 30 and 50 ng/μL,<br> | + | <div align="center"> |
| - | If the concentration is below 30 ng/μL or if 260/280 ratio or 260/230 ratio is | + | <div class="captionedPicture" style="width:50%;float:center;"> |
| + | <a title="Nanodrop" href="https://static.igem.org/mediawiki/2013/f/f0/Nanodrop.png"> | ||
| + | <img alt="Nanodrop" src="https://static.igem.org/mediawiki/2013/f/f0/Nanodrop.png" class="Picture"/> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <b>Figure 1:</b> Figure 1: Nanodrop test after a plasmid purification<br/> | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| + | |||
| + | </div> | ||
| + | |||
| + | |||
| + | <p> | ||
| + | If the concentration is between 30 and 50 ng/μL, concentrate your DNA with SpeedVac concentrator or any other method.<br> | ||
| + | If the concentration is below 30 ng/μL or if 260/280 ratio or 260/230 ratio is not correct, make another purification.<br><br> | ||
| + | |||
| + | <i>260/280 ratio and 260/230 indicate the purity of DNA (or RNA). <br> | ||
| + | Nucleic acids absorb at 260 nm while proteins and phenols absorb at 280 nm and carbohydrates at 230 and other contaminents at 230 nm.<br> | ||
| + | For DNA, a 260/280 ratio around 1,8 is concider to be pure and ange 260/230 ratio must be between 2,0 and 2,2.</i><br></p> | ||
Latest revision as of 13:25, 1 October 2013










Plasmid purification
Goal
The aim of the plasmid purification step is to recover the plasmid produced by the bacteria (cloning strain).
The different step are use to throw other bacterial components off (proteins, cell wall, genomic DNA, etc).
Preparation
Protocol adapted from Macherey-Nagel plasmid purification notebook1. Cell culture
Cultivate cells in LB medium overnight.
2. Cell harvesting
Set saturated E.coli LM culture into 2 mL tubes. Centrifuge at 11 000 x g for 30 secondes. Discard as much as supernatant as possible.
3. Cell lysis
Add 250 μL of Buffer A1 (resuspension buffer). Resuspend the cells with a vortex or a pipette.
Add 250 μL of Buffer A2 (lysis buffer). Mix gently by inverting the tube 6 - 8 times. Incubate at room temperature until lysate appears clear.
Add 300 μL of Buffer A3 (neutralisation buffer). Mix thoroughly by inverting the tube 6 - 8 times .
4. Lysate clarification
Centrifuge at 11 000 x g for 5 minutes . Repeat this step until supernatant is not clear.
5. DNA Binding
Place a NucleoSpin Plasmid Column in a Collection Tube of 2 mL and set the supernatant from the last step. Centrifuge at 11 000 x g for 1 minute. Discard flow-through and place the column back into the collection tube.
6. Membrane washing
Add 600 μL of Buffer A4 (wash buffer) previously supplemented with ethanol. Centrifuge ar 11 000 x g for 1 minute. Discard flow-through and place the column back into an empty collection tube.
7. Dry membrane
Centrifuge at 11 000 x g for 2 minutes and discard the collection tube.
8. DNA Elution
Place the column in a 1,5 mL and add 50 μL de Buffer AE (elution buffer). Incubate at room temperature and centrifuge at 11 000 x g for 1 minute.
Test
Before the storage of the tubes at -20°C, measure the concentration with nanodrop.
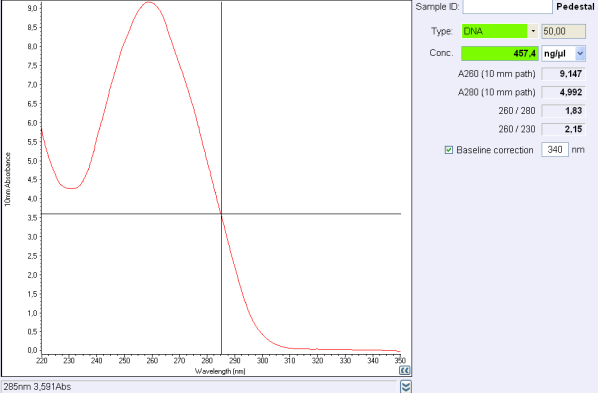
If the concentration is between 30 and 50 ng/μL, concentrate your DNA with SpeedVac concentrator or any other method.
If the concentration is below 30 ng/μL or if 260/280 ratio or 260/230 ratio is not correct, make another purification.
260/280 ratio and 260/230 indicate the purity of DNA (or RNA).
Nucleic acids absorb at 260 nm while proteins and phenols absorb at 280 nm and carbohydrates at 230 and other contaminents at 230 nm.
For DNA, a 260/280 ratio around 1,8 is concider to be pure and ange 260/230 ratio must be between 2,0 and 2,2.


