|
|
| (36 intermediate revisions not shown) |
| Line 1: |
Line 1: |
| - | {{:Team:Evry/template_vo}} | + | {{:Team:Evry/template_protocols}} |
| | + | |
| | <html> | | <html> |
| | | | |
| - | <a href='https://2013.igem.org/Team:Evry/Protocoles/03' title='Vers la page française'> <img src='https://static.igem.org/mediawiki/2013/b/b9/Francais.jpg'/></a> | + | <div id="mainTextcontainer"> |
| | | | |
| - | <h1 align='center'> Protocol for Plasmid purification </h1> | + | <!--<a href='https://2013.igem.org/Team:Evry/Protocoles/03' title='Vers la page française'> <img src='https://static.igem.org/mediawiki/2013/b/b9/Francais.jpg'/></a>--> |
| | | | |
| - | <i>Protocol from Macherey-Nagel plasmid purification handbook</i><br><br> | + | <h1> Plasmid purification </h1> |
| | | | |
| - | A refaire avec MN !
| + | <h2> Goal </h2> |
| | + | <p>The aim of the plasmid purification step is to recover the plasmid produced by the bacteria (cloning strain).<br> |
| | + | The different step are use to throw other bacterial components off (proteins, cell wall, genomic DNA, etc).</p> |
| | | | |
| - | Pre-chill Buffer P3 at 4°C <br><br>
| + | <h2> Preparation </h2> |
| - | <b>1.Pick a single colony</b> from a freshly streaked selective plate and inoculate a starter
| + | |
| - | culture of 2–5 ml LB medium containing the appropriate selective antibiotic. Incubate
| + | |
| - | for approx. 8 h at 37°C with vigorous shaking (approx. 300 rpm).<br>
| + | |
| - | Use a tube or flask with a volume of at least 4 times the volume of the culture.<br><br>
| + | |
| | | | |
| - | <b>2.Dilute the starter culture</b> 1/500 to 1/1000 into selective LB medium. For high-copy | + | <i>Protocol adapted from Macherey-Nagel plasmid purification notebook</i><br><br> |
| - | plasmids, inoculate 25 ml medium with 25–50 μl of starter culture. For low-copy plasmids, inoculate 100 ml medium with 100–200 μl of starter culture. Grow at 37°C for 12–16 h with vigorous shaking (approx. 300 rpm).<br>
| + | |
| - | Use a flask or vessel with a volume of at least 4 times the volume of the culture. The
| + | |
| - | culture should reach a cell density of approximately 3–4 x 109 cells per milliliter,
| + | |
| - | which typically corresponds to a pellet wet weight of approximately 3 g/liter
| + | |
| - | medium.<br><br>
| + | |
| | | | |
| - | <b>3. Harvest the bacterial cells</b> by centrifugation at 6000 x g (6900rpm with rotor AV10/Ser N.0106/04/Cat N.11175754 - Centri CR31 Thermo) for 15 min at 4°C.<br>
| |
| - | If you wish to stop the protocol and continue later, freeze the cell pellets at –20°C.<br><br>
| |
| | | | |
| - | <b>4. Resuspend the bacterial pellet in 4 ml Buffer P1.</b><br> | + | <b><p>1. Cell culture</b><br> |
| - | For efficient lysis it is important to use a vessel that is large enough to allow complete
| + | Cultivate cells in LB medium overnight.<br></p> |
| - | mixing of the lysis buffers. Ensure that RNase A has been added to Buffer P1.If LyseBlue reagent has been added to Buffer P1, vigorously shake the buffer bottle before use to ensure LyseBlue particles are completely resuspended. The bacteria should be resuspended completely by vortexing or pipetting up and down until no
| + | |
| - | cell clumps remain.<br><br>
| + | |
| | | | |
| - | <b>5. Add 4 ml Buffer P2</b>, mix thoroughly by vigorously inverting the sealedtube 4–6 times, and incubate at room temperature (15–25°C) for 5 min.<br>
| |
| - | Do not vortex, as this will result in shearing of genomic DNA. The lysate should
| |
| - | appear viscous. Do not allow the lysis reaction to proceed for more than 5 min. After
| |
| - | use, the bottle containing Buffer P2 should be closed immediately to avoid
| |
| - | acidification from CO2 in the air.<br>
| |
| - | If LyseBlue has been added to Buffer P1 the cell suspension will turn blue after
| |
| - | addition of Buffer P2. Mixing should result in a homogeneously colored suspension.<br>
| |
| - | If the suspension contains localized colorless regions or if brownish cell clumps are
| |
| - | still visible, continue mixing the solution until a homogeneously colored suspension
| |
| - | is achieved.<br><br>
| |
| | | | |
| - | <b>6. Add 4 ml of chilled Buffer P3</b>, mix immediately and thoroughly by | + | <p><b>2. Cell harvesting</b><br> |
| - | vigorously inverting 4–6 times, and incubate on ice for 15 min.<br>
| + | Set saturated E.coli LM culture into 2 mL tubes. Centrifuge at 11 000 x g for 30 secondes. Discard as much as supernatant as possible.<br><p> |
| - | Precipitation is enhanced by using chilled Buffer P3 and incubating on ice. After
| + | |
| - | addition of Buffer P3, a fluffy white material forms and the lysate becomes less
| + | |
| - | viscous. The precipitated material contains genomic DNA, proteins, cell debris, and
| + | |
| - | KDS. The lysate should be mixed thoroughly to ensure even potassium dodecyl sulfate
| + | |
| - | precipitation. If the mixture still appears viscous, more mixing is required to
| + | |
| - | completely neutralize the solution.<br>
| + | |
| - | If LyseBlue reagent has been used, the suspension should be mixed until all trace of
| + | |
| - | blue has gone and the suspension is colorless. A homogeneous colorless suspension
| + | |
| - | indicates that the SDS has been effectively precipitated.<br><br>
| + | |
| | | | |
| - | <b>7. Centrifuge</b> at ≥20,000 x g (10000rpm with rotor AV10/Ser N.0106/04/Cat N.11175754 - Centri CR31 Thermo)for 30 min at 4°C. Remove supernatant containing plasmid | + | <p><b>3. Cell lysis</b> <br> |
| - | DNA promptly.<br>
| + | Add 250 μL of Buffer A1 (resuspension buffer). Resuspend the cells with a vortex or a pipette. |
| - | Before loading the centrifuge, the sample should be mixed again. Centrifugation
| + | <br> |
| - | should be performed in non-glass tubes (e.g., polypropylene). After centrifugation
| + | Add 250 μL of Buffer A2 (lysis buffer). Mix gently by inverting the tube 6 - 8 times. Incubate at room temperature until lysate appears clear.<br> |
| - | the supernatant should be clear.<br> | + | Add 300 μL of Buffer A3 (neutralisation buffer). Mix thoroughly by inverting the tube 6 - 8 times .<br></p> |
| - | Note: Instead of centrifugation steps 7 and 8, the lysate can be efficiently cleared by
| + | |
| - | filtration using a QIAfilter Kits or Cartridges (see www.qiagen.com/products/
| + | |
| - | plasmid/LargeScaleKits).<br><br>
| + | |
| | | | |
| - | <b>8. Centrifuge the supernatant again</b> at ≥20,000 x g (10000rpm with rotor AV10/Ser N.0106/04/Cat N.11175754 - Centri CR31 Thermo) for 15 min at 4°C. Remove | + | <p><b>4. Lysate clarification</b><br> |
| - | supernatant containing plasmid DNA promptly.<br> | + | Centrifuge at 11 000 x g for 5 minutes . Repeat this step until supernatant is not clear.<br></p> |
| - | This second centrifugation step should be carried out to avoid applying suspended
| + | |
| - | or particulate material to the QIAGEN-tip. Suspended material (causing the sample
| + | |
| - | to appear turbid) can clog the QIAGEN-tip and reduce or eliminate gravity flow.<br><br>
| + | |
| | | | |
| - | <b> 9. Equilibrate a QIAGEN-tip 100 </b>by applying 4 ml Buffer QBT, and allow the column to empty by gravity flow.<br> | + | <p><b>5. DNA Binding<br></b> |
| - | Flow of buffer will begin automatically by reduction in surface tension due to the
| + | Place a NucleoSpin Plasmid Column in a Collection Tube of 2 mL and set the supernatant from the last step. Centrifuge at 11 000 x g for 1 minute. Discard flow-through and place the column back into the collection tube.<br></p> |
| - | presence of detergent in the equilibration buffer. Allow the QIAGEN-tip to drain
| + | |
| - | completely. QIAGEN-tips can be left unattended, since the flow of buffer will stop
| + | |
| - | when the meniscus reaches the upper frit in the column.<br><br>
| + | |
| | | | |
| - | <b>10. Apply the supernatant from step 8 to the QIAGEN-tip</b> and allow it to enter the resin | + | <p><b>6. Membrane washing<br></b> |
| - | by gravity flow.<br>
| + | Add 600 μL of Buffer A4 (wash buffer) previously supplemented with ethanol. Centrifuge ar 11 000 x g for 1 minute. Discard flow-through and place the column back into an empty collection tube.<br></p> |
| - | The supernatant should be loaded onto the QIAGEN-tip promptly. If it is left too long
| + | |
| - | and becomes cloudy due to further precipitation of protein, it must be centrifuged | + | |
| - | again or filtered before loading to prevent clogging of the QIAGEN-tip.<br><br>
| + | |
| | | | |
| - | <b>11. Wash the QIAGEN-tip </b>with 2 x 10 ml Buffer QC.<br> | + | <p><b>7. Dry membrane<br></b> |
| - | Allow Buffer QC to move through the QIAGEN-tip by gravity flow. The first wash is
| + | Centrifuge at 11 000 x g for 2 minutes and discard the collection tube.<br></p> |
| - | sufficient to remove all contaminants in the majority of plasmid DNA preparations.
| + | |
| - | The second wash is especially necessary when large culture volumes or bacterial
| + | |
| - | strains producing large amounts of carbohydrates are used.<br><br>
| + | |
| | | | |
| - | <b>12. Elute DNA with 5 ml Buffer QF.</b><br> | + | <p><b>8. DNA Elution<br></b> |
| - | Collect the eluate in a 15 ml or 50 ml tube (not supplied). Use of polycarbonate
| + | Place the column in a 1,5 mL and add 50 μL de Buffer AE (elution buffer). Incubate at room temperature and centrifuge at 11 000 x g for 1 minute.<br></p> |
| - | centrifuge tubes is not recommended as polycarbonate is not resistant to the alcohol | + | |
| - | used in subsequent steps.<br><br>
| + | |
| | | | |
| - | <b>13. Precipitate DNA</b> by adding 3.5 ml (0.7 volume) room-temperature | + | <h2> Test </h2> |
| - | isopropanol to the eluted DNA. Mix and centrifuge immediately at ≥15,000 x g for
| + | <p>Before the storage of the tubes at -20°C, measure the concentration with nanodrop.</p> |
| - | 30 min at 4°C. Carefully decant the supernatant.<br>
| + | |
| - | All solutions should be at room temperature in order to minimize salt precipitation,
| + | |
| - | although centrifugation is carried out at 4°C to prevent overheating of the sample.<br>
| + | |
| - | Alternatively, disposable conical bottom centrifuge tubes can be used for
| + | |
| - | centrifugation at 5000 x g (10000rpm with rotor AV10/Ser N.0106/04/Cat N.11175754 - Centri CR31 Thermo)for 60 min at 4°C. Isopropanol pellets have a glassy
| + | |
| - | appearance and may be more difficult to see than the fluffy, salt-containing pellets
| + | |
| - | that result from ethanol precipitation. Marking the outside of the tube before
| + | |
| - | centrifugation allows the pellet to be more easily located. Isopropanol pellets are also
| + | |
| - | more loosely attached to the side of the tube, and care should be taken when
| + | |
| - | removing the supernatant.<br><br>
| + | |
| | | | |
| - | <b>14. Wash DNA pellet</b> with 2 ml of room-temperature 70% ethanol, and | + | <div align="center"> |
| - | centrifuge at ≥15,000 x g (10000rpm with rotor AV10/Ser N.0106/04/Cat N.11175754 - Centri CR31 Thermo)for 10 min. Carefully decant the supernatant without
| + | <div class="captionedPicture" style="width:50%;float:center;"> |
| - | disturbing the pellet.<br>
| + | <a title="Nanodrop" href="https://static.igem.org/mediawiki/2013/f/f0/Nanodrop.png"> |
| - | The 70% ethanol removes precipitated
| + | <img alt="Nanodrop" src="https://static.igem.org/mediawiki/2013/f/f0/Nanodrop.png" class="Picture"/> |
| - | salt and replaces isopropanol with the more volatile ethanol, making the DNA easier
| + | </a> |
| - | to redissolve.<br><br>
| + | <div class="caption"> |
| | + | <b>Figure 1:</b> Figure 1: Nanodrop test after a plasmid purification<br/> |
| | + | |
| | + | </div> |
| | + | </div> |
| | | | |
| - | <b>15. Air-dry the pellet</b> for 5–10 min, and redissolve the DNA in a suitable volume of buffer
| + | </div> |
| - | (e.g., TE buffer, pH 8.0, or 10 mM Tris·Cl, pH 8.5).<br>
| + | |
| - | Redissolve the DNA pellet by rinsing the walls to recover all the DNA, especially if
| + | |
| - | glass tubes have been used. Pipetting the DNA up and down to promote
| + | |
| - | resuspension may cause shearing and should be avoided. Overdrying the pellet will
| + | |
| - | make the DNA difficult to redissolve. DNA dissolves best under slightly alkaline
| + | |
| - | conditions; it does not easily dissolve in acidic buffers.<br><br>
| + | |
| | | | |
| - | <script type='text/javascript'>writeFooter()</script> | + | |
| | + | <p> |
| | + | If the concentration is between 30 and 50 ng/μL, concentrate your DNA with SpeedVac concentrator or any other method.<br> |
| | + | If the concentration is below 30 ng/μL or if 260/280 ratio or 260/230 ratio is not correct, make another purification.<br><br> |
| | + | |
| | + | <i>260/280 ratio and 260/230 indicate the purity of DNA (or RNA). <br> |
| | + | Nucleic acids absorb at 260 nm while proteins and phenols absorb at 280 nm and carbohydrates at 230 and other contaminents at 230 nm.<br> |
| | + | For DNA, a 260/280 ratio around 1,8 is concider to be pure and ange 260/230 ratio must be between 2,0 and 2,2.</i><br></p> |
| | + | |
| | + | |
| | + | |
| | + | </div> |
| | + | </div> |
| | </html> | | </html> |
| | + | |
| | + | {{:Team:Evry/foot}} |
Plasmid purification
Goal
The aim of the plasmid purification step is to recover the plasmid produced by the bacteria (cloning strain).
The different step are use to throw other bacterial components off (proteins, cell wall, genomic DNA, etc).
Preparation
Protocol adapted from Macherey-Nagel plasmid purification notebook
1. Cell culture
Cultivate cells in LB medium overnight.
2. Cell harvesting
Set saturated E.coli LM culture into 2 mL tubes. Centrifuge at 11 000 x g for 30 secondes. Discard as much as supernatant as possible.
3. Cell lysis
Add 250 μL of Buffer A1 (resuspension buffer). Resuspend the cells with a vortex or a pipette.
Add 250 μL of Buffer A2 (lysis buffer). Mix gently by inverting the tube 6 - 8 times. Incubate at room temperature until lysate appears clear.
Add 300 μL of Buffer A3 (neutralisation buffer). Mix thoroughly by inverting the tube 6 - 8 times .
4. Lysate clarification
Centrifuge at 11 000 x g for 5 minutes . Repeat this step until supernatant is not clear.
5. DNA Binding
Place a NucleoSpin Plasmid Column in a Collection Tube of 2 mL and set the supernatant from the last step. Centrifuge at 11 000 x g for 1 minute. Discard flow-through and place the column back into the collection tube.
6. Membrane washing
Add 600 μL of Buffer A4 (wash buffer) previously supplemented with ethanol. Centrifuge ar 11 000 x g for 1 minute. Discard flow-through and place the column back into an empty collection tube.
7. Dry membrane
Centrifuge at 11 000 x g for 2 minutes and discard the collection tube.
8. DNA Elution
Place the column in a 1,5 mL and add 50 μL de Buffer AE (elution buffer). Incubate at room temperature and centrifuge at 11 000 x g for 1 minute.
Test
Before the storage of the tubes at -20°C, measure the concentration with nanodrop.
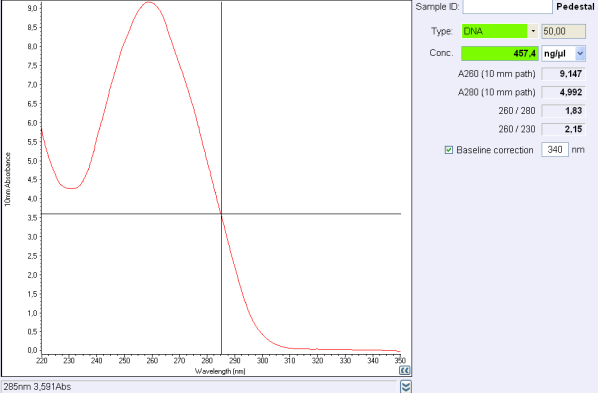
Figure 1: Figure 1: Nanodrop test after a plasmid purification
If the concentration is between 30 and 50 ng/μL, concentrate your DNA with SpeedVac concentrator or any other method.
If the concentration is below 30 ng/μL or if 260/280 ratio or 260/230 ratio is not correct, make another purification.
260/280 ratio and 260/230 indicate the purity of DNA (or RNA).
Nucleic acids absorb at 260 nm while proteins and phenols absorb at 280 nm and carbohydrates at 230 and other contaminents at 230 nm.
For DNA, a 260/280 ratio around 1,8 is concider to be pure and ange 260/230 ratio must be between 2,0 and 2,2.
 "
"












