 "
"
Team:Evry/Protocols/Test
From 2013.igem.org
| (25 intermediate revisions not shown) | |||
| Line 1: | Line 1: | ||
| - | {{:Team:Evry/ | + | {{:Team:Evry/template_notebook}} |
<html> | <html> | ||
| Line 7: | Line 7: | ||
<!--************************** BioBricks Conception overview *********************************************--> | <!--************************** BioBricks Conception overview *********************************************--> | ||
<h1 id="Protocol's_overview">BioBricks conception overview </h1> | <h1 id="Protocol's_overview">BioBricks conception overview </h1> | ||
| + | |||
| + | <!-- Don't touch this area fucking bastard!!!! --> | ||
<div align="center"><img src='https://static.igem.org/mediawiki/2013/2/2d/Overview_of_experiments.png' width="500px" USEMAP="#Link_protocols"/></div> | <div align="center"><img src='https://static.igem.org/mediawiki/2013/2/2d/Overview_of_experiments.png' width="500px" USEMAP="#Link_protocols"/></div> | ||
<MAP NAME="Link_protocols"> | <MAP NAME="Link_protocols"> | ||
| - | <AREA SHAPE="rect" COORDS="0,5, | + | <AREA SHAPE="rect" COORDS="0,5,104,76" HREF="#Media"></AREA> |
| - | <AREA SHAPE="rect" COORDS=" | + | <AREA SHAPE="rect" COORDS="172,5,277,76" HREF="#DNA_genomic_extraction"></AREA> |
| - | <AREA SHAPE="rect" COORDS=" | + | <AREA SHAPE="rect" COORDS="28,127,238,179" HREF="#Polymerase_Chain_Reaction"></AREA> |
| - | <AREA SHAPE="rect" COORDS=" | + | <AREA SHAPE="rect" COORDS="29,223,238,251" HREF="#DNA_genomic_extraction"></AREA> |
| - | <AREA | + | <AREA SHAPE="rect" COORDS="200,292,340,330" HREF="#Electrophoresis"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | <AREA SHAPE="rect" COORDS="29,380,238,410" HREF="#Golden_Gate"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | <AREA SHAPE="rect" COORDS="292,380,500,410" HREF="#Competent_cells"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | <AREA SHAPE="rect" COORDS="148,480,356,509" HREF="#Transformation"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | <AREA SHAPE="rect" COORDS="148,545,356,574" HREF="#Streaking"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | <AREA SHAPE="rect" COORDS="318,630,465,669" HREF="#Electrophoresis"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | <AREA SHAPE="rect" COORDS="148,725,356,755" HREF="#Plasmid_purification"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | <AREA SHAPE="rect" COORDS="318,810,465,850" HREF="#Sequencing"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | <AREA SHAPE="rect" COORDS="148,903,356,933" HREF="#Transformation"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | <AREA SHAPE="rect" COORDS="148,962,356,990" HREF="#BioBrick_Assembly"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | <AREA SHAPE="rect" COORDS="148,1027,356,1057" HREF="#Characterization_overview"></AREA> |
| - | <AREA SHAPE="rect" COORDS="" HREF="#"> | + | |
</MAP> | </MAP> | ||
| - | < | + | <!-- You can edit after this comment --> |
| - | + | ||
| + | <br id="Media"/> | ||
<br/> | <br/> | ||
<!--************************* Solutions and Media **********************************************--> | <!--************************* Solutions and Media **********************************************--> | ||
| - | <h1 | + | <h1 > Solutions, Media and Petri Dishes </h1> |
<h2> Antibiotics </h2> | <h2> Antibiotics </h2> | ||
| Line 239: | Line 240: | ||
<i>Tris HCl is used to store DNA.</i></p> | <i>Tris HCl is used to store DNA.</i></p> | ||
| + | |||
| + | <br id="DNA_genomic_extraction"/> | ||
<br/> | <br/> | ||
| - | |||
| - | |||
<!--********************************* DNA genomic extraction ********************************************--> | <!--********************************* DNA genomic extraction ********************************************--> | ||
| - | <h1 | + | <h1 >DNA genomic extraction</h1> |
| Line 367: | Line 368: | ||
<br/> | <br/> | ||
| - | <!--*************************************** Golden Gate | + | <!--*************************************** Golden Gate *****************************************************--> |
<h1 > Golden Gate</h1> | <h1 > Golden Gate</h1> | ||
| Line 393: | Line 394: | ||
<br/> | <br/> | ||
| - | <!-- | + | <!--************************************* Competent cells ******************************************************--> |
<h1 > Competent cells </h1> | <h1 > Competent cells </h1> | ||
| Line 478: | Line 479: | ||
<br/> | <br/> | ||
| - | <!--*************************************** Transformation | + | <!--*************************************** Transformation **************************************************--> |
<h1 > Transformation </h1> | <h1 > Transformation </h1> | ||
| Line 505: | Line 506: | ||
| - | <!-- | + | <!--************************************** Plasmid purification *********************************************--> |
<h1> Plasmid purification </h1> | <h1> Plasmid purification </h1> | ||
| Line 555: | Line 556: | ||
<div class="caption"> | <div class="caption"> | ||
<b>Figure 1:</b> Figure 1: Nanodrop test after a plasmid purification<br/> | <b>Figure 1:</b> Figure 1: Nanodrop test after a plasmid purification<br/> | ||
| - | |||
</div> | </div> | ||
| - | + | </div> | |
| + | |||
| + | <div style="clear:both"></div> | ||
| Line 570: | Line 572: | ||
<br/> | <br/> | ||
| - | <!-- | + | <!--**************************** Gel electrophoresis analysis ********************************************--> |
<h1> Gel electrophoresis analysis</h1> | <h1> Gel electrophoresis analysis</h1> | ||
| Line 592: | Line 594: | ||
<br/> | <br/> | ||
| - | <!-- | + | <!--********************************** PCR Purification *********************************************--> |
<h1> PCR Purification </h1> | <h1> PCR Purification </h1> | ||
| Line 633: | Line 635: | ||
| - | <!-- | + | <!--**************************** BioBrick Assembly ********************************************--> |
<h1 > BioBrick Assembly </h1> | <h1 > BioBrick Assembly </h1> | ||
| Line 809: | Line 811: | ||
| - | <!-- | + | <!--******************************** BioBricks characterization *************************************************--> |
<h1 >BioBricks Characterization overview</h1> | <h1 >BioBricks Characterization overview</h1> | ||
| + | |||
| + | <div align="center"><img src='https://static.igem.org/mediawiki/2013/d/d2/Caracterisation.png' width="500px" USEMAP="#Link_Characterization"/></div> | ||
<br id="TECAN_Analysis" /> | <br id="TECAN_Analysis" /> | ||
| Line 816: | Line 820: | ||
| - | <!-- | + | <!--********************************** TECAN Analysis ***************************************************--> |
<h1 >TECAN Analysis</h1> | <h1 >TECAN Analysis</h1> | ||
| + | |||
| + | <h2> Goal </h2> | ||
| + | <p> | ||
| + | The Tecan is a plate reader that is able to characterize our bacteria of interest by measuring, for example, growth rate (OD) and/or protein production (GFP). The software saves the measurements over time, thus allowing to study the kinetics of the studied phenomena. With a 96 wells plate (8x12), we are able to test various conditions. | ||
| + | </p> | ||
| + | |||
| + | <h2> Steps </h2> | ||
| + | <p> | ||
| + | The hardest part when you use a plate reader to characterize your parts is to carefully choose your controls and anticipate the timings of your bacterial pre-cultures.</br> | ||
| + | </br> | ||
| + | We tested various controls during this summer:</br> | ||
| + | <ul> | ||
| + | <li>TOP10 growth control</br> | ||
| + | <li>BL21 growth controlFor GFP expression: GFP expression under the control of a pLac promoteLac operator in a delta-LacI strain and GFP expression under the control of a Fur operator in a Δ-Fur strain. | ||
| + | </ul> | ||
| + | </p> | ||
| + | |||
| + | <h2> Preparation </h2> | ||
| + | <h3>Medium preparation</h3> | ||
| + | |||
| + | <p> | ||
| + | LB medium emit a side signal. As turbidity (OD) and fluorescence of our sample are measured, M9 medium is use instead. | ||
| + | |||
| + | |||
| + | </p> | ||
| + | |||
| + | <p> | ||
| + | <u>Composition for 50 mL of:</u> | ||
| + | </p> | ||
| + | |||
| + | <table cellpadding="10" cellspacing="0" align='center' border="1"> | ||
| + | <thead> | ||
| + | <tr> | ||
| + | <th align="center"> | ||
| + | Reagent | ||
| + | </th> | ||
| + | <th align="center"> | ||
| + | M9 medium (without iron) | ||
| + | </th> | ||
| + | <th align="center"> | ||
| + | M9 medium (with iron) | ||
| + | </th> | ||
| + | </tr> | ||
| + | </thead> | ||
| + | |||
| + | <tbody> | ||
| + | <tr> | ||
| + | <td align='center'> | ||
| + | M9 salt (5X) | ||
| + | </td> | ||
| + | <td colspan="2" align='center'> | ||
| + | 10 mL | ||
| + | </td> | ||
| + | </tr> | ||
| + | |||
| + | <tr> | ||
| + | <td align='center'> | ||
| + | CaCl<sub>2</sub> (1M) | ||
| + | </td> | ||
| + | <td colspan="2" align='center'> | ||
| + | 5 µL | ||
| + | </td> | ||
| + | </tr> | ||
| + | |||
| + | <tr> | ||
| + | <td align='center'> | ||
| + | MgSO<sub>4</sub> (1M) | ||
| + | </td> | ||
| + | <td colspan="2" align='center'> | ||
| + | 100 µL | ||
| + | </td> | ||
| + | </tr> | ||
| + | |||
| + | <tr> | ||
| + | <td align='center'> | ||
| + | Glycerol (50%) | ||
| + | </td> | ||
| + | <td colspan="2" align='center'> | ||
| + | 800 µL | ||
| + | </td> | ||
| + | </tr> | ||
| + | |||
| + | <tr> | ||
| + | <td align='center'> | ||
| + | Thiamine | ||
| + | </td> | ||
| + | <td colspan="2" align='center'> | ||
| + | 5 µL | ||
| + | </td> | ||
| + | </tr> | ||
| + | |||
| + | <tr> | ||
| + | <td align='center'> | ||
| + | NaOH (pH 7.4) | ||
| + | </td> | ||
| + | <td colspan="2" align='center'> | ||
| + | 12.5 µL | ||
| + | </td> | ||
| + | </tr> | ||
| + | |||
| + | <tr> | ||
| + | <td align='center'> | ||
| + | H<sub>2</sub>O | ||
| + | </td> | ||
| + | <td align='center'> | ||
| + | 40 mL | ||
| + | </td> | ||
| + | <td align='center'> | ||
| + | 39 mL | ||
| + | </td> | ||
| + | </tr> | ||
| + | |||
| + | <tr> | ||
| + | <td align='center'> | ||
| + | FeSO<sub>4</sub> (10mM) | ||
| + | </td> | ||
| + | <td align='center'> | ||
| + | - | ||
| + | </td> | ||
| + | <td align='center'> | ||
| + | 50 µL | ||
| + | </td> | ||
| + | </tr> | ||
| + | |||
| + | <tr> | ||
| + | <td align='center'> | ||
| + | Casamino acids (0.2%) | ||
| + | </td> | ||
| + | <td align='center'> | ||
| + | - | ||
| + | </td> | ||
| + | <td align='center'> | ||
| + | 1 mL | ||
| + | </td> | ||
| + | </tr> | ||
| + | </tbody> | ||
| + | </table> | ||
| + | |||
| + | <p><br> | ||
| + | Once the mixture is prepared, the medium must be filtered to be sterilised using 0.22 µm filter. | ||
| + | </p> | ||
| + | |||
| + | <h3>Pre-culture preparation</h3> | ||
| + | |||
| + | <p> | ||
| + | In a 15 mL tube, add 2 mL of M9 medium and inoculate BL21 cells (expression strain) from glycerol<br/> | ||
| + | To inhibit the expression of sfGPF, use M9 with iron, instead of classical M9. | ||
| + | <br/> | ||
| + | After one night of culture, refresh the precultures by diluting them 200 times in M9 medium (with iron and carbenicillin). | ||
| + | After 8 hours of culture, prepare your wells plate according to the following scheme:<br/></p> | ||
| + | |||
| + | |||
| + | <div align="center"><img src=' | ||
| + | https://static.igem.org/mediawiki/2013/f/f8/Tecan.png' width="500px"/></div> | ||
| + | <p><i> | ||
| + | Technical triplicate are made to see the variation induced by the manipulator.<br/> | ||
| + | Biological duplicate are made to determine the natural variation of the process observed.</i> | ||
| + | <br/></p> | ||
| + | |||
| + | |||
<br id="Microfluidic_experiment" /> | <br id="Microfluidic_experiment" /> | ||
<br/> | <br/> | ||
| - | <!-- | + | <!--************************************ Microfluidic experiment *********************************************--> |
<h1 >Microfluidic experiment </h1> | <h1 >Microfluidic experiment </h1> | ||
| + | <h2> Goal </h2> | ||
| + | <p>The aim of microfluidic analysis is to trap bacteria in a chip, with a continuous medium. Hence, the behaviour (e.g. GFP production) of bacteria in exponnential phase can be analyse with microscope during long acquisition (>24h).</p> | ||
| + | |||
| + | <h2> Chip preparation </h2> | ||
| + | |||
| + | <div class="captionedPicture" style="width:25%;float:right;"> | ||
| + | <a title="Chip Mould" href="https://static.igem.org/mediawiki/2013/thumb/6/6c/Microfluidic.JPG/561px-Microfluidic.JPG"> | ||
| + | <img alt="Chip Mould" src="https://static.igem.org/mediawiki/2013/thumb/6/6c/Microfluidic.JPG/561px-Microfluidic.JPG" class="Picture"/> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <b>Figure 1:</b> Microfluidic ship mould<br/> | ||
| + | <i>(designed by Boris Kirov)</i> | ||
| + | </div> | ||
| + | </div> | ||
| + | |||
| + | <p>Mix gently 36 g of Polydiméthylsiloxane (PDMS,condensed formula: (C<inf><small>2</inf></small>H<inf><small>6</inf></small>OSi)<inf><small>n</inf></small> ) with 3.6 g of curring agent (which make the PDMS polymerize).<br/> | ||
| + | Put in a 50 mL tube and centifugate it at 13 000 for 1 minute to remove bubbles from the mixture.<br/> | ||
| + | Pour the mixture on the chip mould and put it under a vacuum pump.<br/> | ||
| + | |||
| + | |||
| + | |||
| + | |||
| + | Once there is no more bubbles that go out of the mixture, put it in the oven at 80°C for 2 hours.<br/> | ||
| + | Separate the different chips and make the holes of the input and outpout of channels with a punch.<br/> | ||
| + | Remove all the dust on chips surfaces. Then put chips and glass slides on plasma oven.<br/> | ||
| + | After that, put quickly the chip on glass slide and put them in the oven at 80°C for 2 hours.<br/> | ||
| + | |||
| + | <h2> Bacteria preparation </h2> | ||
| + | <p>Make 30 mL of preculture of your strain of interest in LB medium with the recquired antibiotics to avoid contamination. <br/> | ||
| + | Let it growth overnight.<br/> | ||
| + | Centrifugate and resuspend the pellet in 3 mL of LB medium with the recquired antibiotics and mild detergent<br/> | ||
| + | Inject bacteria in the chip with needle and syringe. | ||
| + | |||
| + | |||
| + | <div align="center"> | ||
| + | <div class="captionedPicture" style="width:85%;float:center;"> | ||
| + | <a title="It's a trap !" href="https://static.igem.org/mediawiki/2013/thumb/8/87/Microfluidic_acquisition.png/800px-Microfluidic_acquisition.png"> | ||
| + | <img alt="It's a trap !" src="https://static.igem.org/mediawiki/2013/thumb/8/87/Microfluidic_acquisition.png/800px-Microfluidic_acquisition.png" class="Picture"/> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <b>Figure 2:</b> Microfluidic system acquisition from microfluidic ship (A: two ship), microscop (B) and injecting system (C) | ||
| + | </div> | ||
| + | </div> | ||
| + | |||
| + | </div> | ||
| + | <br/> | ||
| + | |||
| + | |||
| + | Prepare 2x25 mL of LB medium with antibiotic and mild detergent and inject it in the chip.<br/> | ||
| + | Check your preparation with the microscope and let the bacteria growth overnight.</br></p> | ||
| + | |||
| + | <div align="center"> | ||
| + | <div class="captionedPicture" style="width:40%;float:center;"> | ||
| + | <a title="It's a trap !" href="https://static.igem.org/mediawiki/2013/thumb/2/23/SNAP-174209-0004.png/709px-SNAP-174209-0004.png"> | ||
| + | <img alt="It's a trap !" src="https://static.igem.org/mediawiki/2013/thumb/2/23/SNAP-174209-0004.png/709px-SNAP-174209-0004.png" class="Picture"/> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <b>Figure 3:</b> A <i>Escherichia coli</i> (TOP 10 strain) in a microfluidic trap.<br/> | ||
| + | At the top and bottom of the picture, the flow of medium go throught the 2 channels. | ||
| + | </div> | ||
| + | </div> | ||
| + | |||
| + | </div> | ||
| + | |||
| + | <h2>Acquisition</h2> | ||
| + | <p>Wash the chip with your recquired medium (in our case: M9 with or without iron).<br/> | ||
| + | <p> | ||
| + | |||
<br id="Homologous_recombination" /> | <br id="Homologous_recombination" /> | ||
<br/> | <br/> | ||
| - | <!-- | + | <!--*************************** ΔFur strain by homologous recombination ***********************************--> |
<h1 >Homologous recombination </h1> | <h1 >Homologous recombination </h1> | ||
| Line 892: | Line 1,124: | ||
<br>Streak plate on LB plates and grow over night at <b>42°C</b>. | <br>Streak plate on LB plates and grow over night at <b>42°C</b>. | ||
| - | <br> | + | <br>Repeat the grid. |
| - | + | ||
| - | + | ||
</p> | </p> | ||
| Line 965: | Line 1,195: | ||
<h3> Bacteria </h3> | <h3> Bacteria </h3> | ||
| + | |||
| + | <div align="center"> | ||
| + | <div class="captionedPicture" style="width:40%;float:center;"> | ||
| + | <a title="Siderophores" href="https://static.igem.org/mediawiki/2013/4/46/Siderophore_detection.png"> | ||
| + | <img alt="Siderophores" src="https://static.igem.org/mediawiki/2013/4/46/Siderophore_detection.png" class="Picture"/> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <b>Figure 1:</b> CAS plate with four different bacteria plated. Moving clockwise from top left: <i>Micrococcus luteus</i>, <i>Staphylococcus aureus</i>, <i>Enterobacter aerogenes</i>, <i>Escherichia coli</i> | ||
| + | |||
| + | </div> | ||
| + | </div> | ||
| + | </div> | ||
<div id="citation_box"> | <div id="citation_box"> | ||
Latest revision as of 02:35, 5 October 2013










BioBricks conception overview

Solutions, Media and Petri Dishes
Antibiotics
Carbenicillin:
Put 150 mg of Carbenicilline in 15 mL of distilled water (10 mg per mL).
Filter with a 0,22 µm millipore,divide in 2 mL tubes and store at -4 °C.
Use 1 µL of Carbenicillin for 1 mL of solution.
Chloramphenicol:
Put 525 mg of Kanamycin in 15 mL of ethanol (35 mg per mL).
Filter with a 0,22 µm millipore,divide in 2 mL tubes and store at -4 °C.
Use 25 µL of Chloramphenicol for 1 mL of solution.
Kanamycin:
Put 750 mg of Kanamycin in 15 mL of distilled water (50 mg per mL).
Filter with a 0,22 µm millipore,divide in 2 mL tubes and store at -4 °C.
Use 1 µL of Kanamycin for 1 mL of solution.
Spectinomycin:
Put 900 mg of Spectinomycin in 15 mL of ( 60 mg per mL).
Filter with a 0,22 µm millipore,divide in 2 mL tubes and store at -4°C.
Use 1 µL of Spectinomycin for 1 mL of solution.
Thoses antibiotics are used to select bacteria with a plasmid including an interest construction and the corresponding antibiotic resistance gene.
LB and LB Agar
Mix 20 g of LB Broth in 1L of distilled water in a bottle of 1L, then put in autoclave.
Mix 17,5 g of LB Agar in 500 mL of distilled water in a bottle of 1L, then put in autoclave.
M9 Medium
Composition for 50 mL of:
| Reagent | M9 medium (without iron) | M9 medium (with iron) |
|---|---|---|
| M9 salt (5X) | 10 mL | |
| CaCl2 (1M) | 5 µL | |
| MgSO4 (1M) | 100 µL | |
| Glycerol (50%) | 800 µL | |
| Thiamine | 5 µL | |
| NaOH (pH 7.4) | 12.5 µL | |
| H2O | 40 mL | 39 mL |
| FeSO4 (10mM) | - | 50 µL |
| Casamino acids (0.2%) | - | 1 mL |
Once the mixture is prepared, the medium must be filtered to be sterilised using 0.22 µm filter.
Petri dish
Melt the LB Agar in a microwave, and split it in 50 mL tubes.
When the tubes have cooled, put the necessary amount of antibiotics in it (50 µL for Carbenicillin and Kanamycin, 1250 µL for Chloramphenicol) and mix thoroughly.
Put 25 mL of the solution in a Petri dish.
CaCl2 and CaCl2+glycerol
Solution for 1M Cacl2:
Add 14,30g of CaCl2 into 100 ml desalted water
Solution for 0,1M Cacl2:
Add 50 mL of CaCl2 1M solution into 450 ml of desalted water
Solution for 0,1M Cacl2 + 15% glycerol:
Add 50 mL of CaCl2 1M solution and 75 mL of glycerol 100% into 450 ml of desalted water
Tris-Acetate-EDTA (TAE) Buffer solutions
NaOH solution 5M 100 mL:
Add 19,99 g of NaOH in 100 mL of distilled water.
EthyleneDiamineTetraacetic Acid (EDTA) solution 0,5M 300 mL:
Add 43,8 g of EDTA in 250 mL of distilled water.
Add NaOH 5M until EDTA is solubilized and pH~8.
Add distilled water until you reach 300 mL.
TAE Stock solution 50X 300 mL:
Add 60,5 g of TAE in 187,5 mL of distilled water.
Add 14,27 mL of acetate solution (previously put at 4°C).
Add 25 mL of EDTA solution previously prepared.
Add distilled water until you reach 250 mL.
TAE solution 1X 100 mL:
Add 5 mL of TAE Stock solution 50X in 100 mL of distilled water.
Tris HCl
Disolve 4,6g of Trisbase in 30 mL of distilled water.
Adjust pH with concentrated HCl(~4 mL) until pH=7,5.
Add distilled water until 50 mL then put in autoclave.
If the solution has a yellow coloration, do the preparation again with better Trisbase.
Tris HCl is used to store DNA.
DNA genomic extraction
Goal
The aim of the extraction step is to recover Escherichia coli genomic DNA.
The different step are use to throw other bacterial components off (proteins, cell wall, plasmids,etc).
Once we have E.coli genomic DNA
Preparation
Protocol adapted from Thermo Scientific Genomic extraction notebook1. Cell culture
Cultivate cells in LB medium overnight.
2. Cell harvesting
Set saturated E.coli LM culture into 2 mL tubes. Centrifuge at 8 000 x g for 5 minutes. Discard as much as supernatant as possible.
3. Cell lysis
Add 180 μL of Digestion Solution and 20 μL of Proteinase K Solution. Resuspend the cells thoroughly with a vortex or a pipette.
Incubate the tubes at 56°C while vortexing occassionally until the cells are completely lysed(~30 minutes).
Add 20 μL of RNase A solution, mix by vortexing and incubate the tubes for 10 minutes at room temperature.
Add 200 μL of Lysis Solution to the sample. Mix thoroughly by vortexing until a homogeneous mixture is obtained. (~15 secondes)
Add 400 μL of 50% ethanol and mix with a vortex or a pipette.
4. DNA Binding
Transfer the lysate to a GeneJET Genomic DNA Purification Column inserted in a collection tube. Centrifuge the column at 12 000 x g for 1 minute.
Discard the collection tube containing flow-through solution and place the column into a new 2 mL tube.
5. Membrane washing
Add 500 μL of Wash Buffer I (previously added with ethanol). Centrifuge at 1 600 x g. Discard the flow-through and place the purification column back into the collection tube.
Add 500 μL of Wash Buffer II (previously added with ethanol)to the purification column.
6. Dry membrane
Centrifuge at 16 000 x g for 3 minutes.
7. DNA Elution
Add 200 μL of Elution Buffer to the center of the purification column membrane to elute genomic DNA. Incubate for 2 minutes at room temperature and centrifuge at 8 000 x g for 1 minute.
Discard the purification column, test the concentration with a Nanodrop.
Store the purified DNA in TrisHCl at -20°C or use it immediatly.
Polymerase Chain Reaction
Principle

Polymerase Chain Reaction (PCR) is a molecular biology method used to amplify a small amount of genetic material (DNA or RNA), using specific primers of a target sequence.
PCR is divided into 5 steps:
- First denaturation:
- Denaturation step
- Annealing step
- Elongation step
- Final step
Denaturation step occure between 94 and 98°C.The heat breaks the hydrogen bonds and then causes the separation of the double strand of DNA into two single strands.
Annealing step occure between 50 and 65 °C. Primers anneals to DNA simple strand by complementarity of the nitrogenized bases.
Elongation step occure between 70 and 80°C, depending on the DNA polymerase used. The polymerase synthesizes a new DNA strand complementary to the DNA template
Preparation
Mixture preparation
We use two differents polymerase for our PCR: Taq Polymerase and Q5.
As Q5 is a hight fidelity polymerase, we use it to obtain sequences without mutation (for a total of 50 μL of mixture); otherwise we use Taq Polymerase instead (for a total of 25 μL of mixture).
Afterwards, we will describe the protocol for a PCR with Q5 (total amount: 50 μL).
Note: Proportion are the same with Taq Polymerase, for 25 μL or 50 μL: 1:10 of each primer, 1:5 of Buffer, 1:50 of dNTPs, 1:50 of genomic DNA and 1:100 of polymerase
Make tubes with 5 μL (1:10) of Forward primer and 5 μL (1:10) of Reverse primer for each desidered sequence.
For positive and negative controle, use primers that worked previously in the same condition of PCR.
We use primers (Primers 009 and 010) for pEntC for ou controles.
For more details about our primers, see the corresponding page.
For n tubes, prepare a master mix with the following solutions:
- (n+1) x 10 μL of Q5 Buffer
- (n+1) x 1 μL of dNTPs
- (n+1) x 27,5 μL of distilled water.
- (n+1) x 0,5 μL of Q5
Note: Take the enzyme out of -20°C just before utilisation.
Split 39 μL of the master mix in each tube.
Add 1 μL of genomic DNA in each tube, except in your negative controle tube. In this one, put 1 μL of distilled water instead.
PCR Cycles
Set the following program on your Thermo Cycler:
- First denaturation: 30 seconds at 98°C
- Denaturation step 10 seconds at 98°C
- Annealing step 20 seconds at 56°C
- Elongation step (n+1) minutes at 72°C (as n is the number of kB of your sequence)
- Final step 5 minutes at 72°C
Steps 2, 3 and 4 are repeated 29 times.
Then let the mixture cool down at 4°C.
Note: Annealing temperature depend on your primers and elongation temperature on the polymerase used.
Golden Gate
Goal
The aim of Golden Gate assembly is to assemble in one step various fragment of DNA.
Preparation
Competent cells
Goal
Bacteria can integrate DNA fragments from the environment. Escherichia coli, contrary to Bacillus subtillis another bacteria frequently used in molecular and synthetic biology, is not naturally in a state of competence (able to integrate DNA fragments during transformation), that is why we have to prepare them.
Two methods can be used:
-Chimiocompetent cells
-Electrocompetent cells
We choose to use chimiocompetent cells for usual transformation and electrocompetent cells just for ΔFur strain preparation by homologous recombination.
Preparation
Grow overnight pre-culture of the strain you want to make competent (chimio or electro) in 2 mL of LB.
Chimiocompetent cells
Solutions preparation
Solution for 1M Cacl2:
Add 14,30g of CaCl2 into 100 ml distilledwater
Solution for 0,1M Cacl2:
Add 50 mL of CaCl2 1M solution into 450 ml of distilled water
Solution for 0,1M Cacl2 + 15% glycerol:
Add 50 mL of CaCl2 1M solution and 75 mL of glycerol 100% into 450 ml of dissilted water
Competent cells preparation
For 200 ml LB medium, add 400 µL of strain sample.
Let the bacteria grow until it reaches an Optical Density (OD) between 0,3 and 0,35. Chimiocompetent bacteria are more able to transform when they are in exponential phase.
Once it reached the right OD, put the medium on ice for 30 minutes to slow down growth.
Split the 200 mL into 4x50 mL tubes then centrifuge at 3000 rpm for 5 minutes at 4°C and suppress supernatant afterwards.
Resuspend the pellet cells with 5 mL of Cacl2 at 0,1M for each 50 mL tube.
Again, put the medium on ice for 30 minutes.
Centrifuge at 3000 rpm for 5 minutes at 4°C then suppress supernatant.
Resuspend the cells with 1 mL of Cacl2 at 0,1M + 15% glycerol for each 50 mL tube.
Split the total 4 mL into 40 tubes containing each 100 µL of concentrated cell solution. This step should be executed fast enough and on ice.
Store at - 80°C.
Electrocompetent cells
For 200 ml LB medium, add 200 µL of strain sample.
Let the bacteria grow until it reaches an OD between 0,5 and 0,7.
Once it reached the right OD, put the medium on ice for 20 minutes to slow down growth.
Split the 200 mL into 4x50 mL tubes then centrifuge at 4000 rpm for 15 minutes at 4°C and suppress supernatant afterwards.
Resuspend the pellet cells with 50 mL of ice-cold 10% glycerol for each 50 mL tube.
Centrifuge at 4000 rpm for 15 minutes at 4°C then suppress supernatant.
Resuspend the cells with 25 mL of ice-cold 10% glycerol for each 50 mL tube.
Centrifuge at 4000 rpm for 15 minutes at 4°C then suppress supernatant.
Resuspend the cells with 2 mL of ice-cold 10% glycerol for each 50 mL tube.
Centrifuge at 4000 rpm for 15 minutes at 4°C then suppress supernatant.
Resuspend the cells with 2 mL of ice-cold 10% glycerol for each 50 mL tube.
Split the total 4 mL into 40 tubes containing each 100 µL of concentrated cell solution. This step should be executed fast enough and on ice.
Store at - 80°C.
Contamination and competence tests
Plate each strain on LB medium with Ampicillin, Kanamycin or Chloramphenicol in order to evaluate if it contaminated or not.
Transform each strain with a pSB1A3 plasmid (red colonies) and plated them on LB medium with Ampicillin only to evaluate wether our strains are competent or not.
If the bacteria is competent, it will incorporate the plasmid that code for a red protein and thus have a red phenotype.(see figure below)
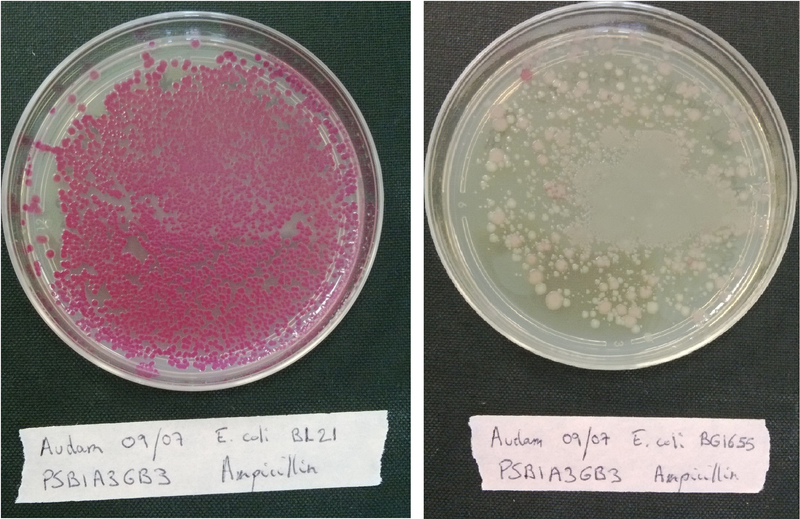
Transformation
Goal
In this step, the heat shock (between -80°C up to 42°C) induces chimiocompetent bacteria to be in state of competence. DNA from the environment (in this case: plasmid) can then integrate the cell.
Preparation
Chimiocompetent cells
For 100 µL of chemical competent cells, add 1 µL of plasmidic DNA. If the ligation protocol is a
Golden Gate , add 5 µL instead.
Let 30 minutes on ice.
Let the cells at 42°C for exactly 50 seconds.
Let 5 minutes on ice.
Resuspend the cells with 1 mL of LB spread on a petri dish then let it at 37°C.
Controles
To check if the transformation have work, make a positive and a negative controle.
If the transformation have worked, colonies must have a red phenotype.
Without plasmid with antibiotic resistance, no colonies must grow.
Plasmid purification
Goal
The aim of the plasmid purification step is to recover the plasmid produced by the bacteria (cloning strain).
The different step are use to throw other bacterial components off (proteins, cell wall, genomic DNA, etc).
Preparation
Protocol adapted from Macherey-Nagel plasmid purification notebook1. Cell culture
Cultivate cells in LB medium overnight.
2. Cell harvesting
Set saturated E.coli LM culture into 2 mL tubes. Centrifuge at 11 000 x g for 30 secondes. Discard as much as supernatant as possible.
3. Cell lysis
Add 250 μL of Buffer A1 (resuspension buffer). Resuspend the cells with a vortex or a pipette.
Add 250 μL of Buffer A2 (lysis buffer). Mix gently by inverting the tube 6 - 8 times. Incubate at room temperature until lysate appears clear.
Add 300 μL of Buffer A3 (neutralisation buffer). Mix thoroughly by inverting the tube 6 - 8 times .
4. Lysate clarification
Centrifuge at 11 000 x g for 5 minutes . Repeat this step until supernatant is not clear.
5. DNA Binding
Place a NucleoSpin Plasmid Column in a Collection Tube of 2 mL and set the supernatant from the last step. Centrifuge at 11 000 x g for 1 minute. Discard flow-through and place the column back into the collection tube.
6. Membrane washing
Add 600 μL of Buffer A4 (wash buffer) previously supplemented with ethanol. Centrifuge ar 11 000 x g for 1 minute. Discard flow-through and place the column back into an empty collection tube.
7. Dry membrane
Centrifuge at 11 000 x g for 2 minutes and discard the collection tube.
8. DNA Elution
Place the column in a 1,5 mL and add 50 μL de Buffer AE (elution buffer). Incubate at room temperature and centrifuge at 11 000 x g for 1 minute.
Test
Before the storage of the tubes at -20°C, measure the concentration with nanodrop.
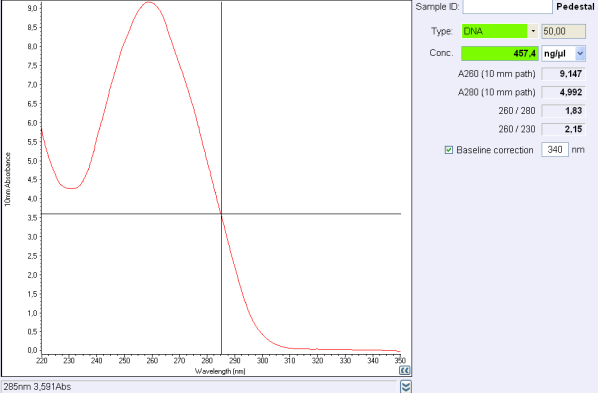
If the concentration is between 30 and 50 ng/μL, concentrate your DNA with SpeedVac concentrator or any other method.
If the concentration is below 30 ng/μL or if 260/280 ratio or 260/230 ratio is not correct, make another purification.
260/280 ratio and 260/230 indicate the purity of DNA (or RNA).
Nucleic acids absorb at 260 nm while proteins and phenols absorb at 280 nm and carbohydrates at 230 and other contaminents at 230 nm.
For DNA, a 260/280 ratio around 1,8 is concider to be pure and ange 260/230 ratio must be between 2,0 and 2,2.
Gel electrophoresis analysis
Principle
Electrophoresis is a method using electrical field to separate DNA or RNA sequence by size. Smaller the fragment is, the more it migrates on the gel.
Using a DNA ladder, we can know the size of DNA sequence and then check if we have the sequence we wanted.
Preparation
Put 0.5 g of Agar in 50 mL of TAE 1X and heat until the mix is melted.
Once the agar have dissolved and the mixture have cooled down, put 0.3 mL of EtBr in it.
Pour the mixture on the mould, put the comb and wait until the gel polymerized.
Put the gel in the electrophoresis unit (with TAE 1X).
Put your DNA sample (with loading dye) in the PCR well, do not forget the kB ladder.
After the migration (~30 to 45 minutes depanding to your fragment size), observe the gel with UV light.
PCR Purification
Goal
The aim of the PCR purification step is to recover DNA from PCR and other enzymatic reaction mixtures.
Preparation
Protocol adapted from Thermo Scientific PCR Purification notebook1. Mixture preparation
Add 1:1 volume of Binding Buffer to completed PCR mixture (e.g. if you have 100 µL of reaction mixture, add 100 µL of Binding Buffer).
Mix thoroughly and check the solution's color: a yellow color color indicates an optimal pH for DNA binding.
If the color of the solution is orange or violet, add 10 µL of 3 M sodium acetate, pH 5,2 and mix. The color will become yellow.
If the DNA fragment is superior to 500 bp, add a 1:2 volume of 100% isopropanol (e.g. if you have 100 µL of PCR mixture combined with 100 µL (total amount of 200 µL), add 100 µL isopropanol). Mix thoroughly.
2. Binding DNA
Transfer up 800 µL of the solution from step previous step to the GeneJET purification column.
Centrifuge for 30-60 seconds, then discard the flow-through.
3. Membrane washing
Add 700 µL of Wash Buffer (previously dilted with ethanol) to the purification column.
Centrifuge for 30-60 seconds, then discard the flow-through and place the purification column back into the collection tube?
4. Dry membrane
Centrifuge the empty GeneJET purification for 1 minute to completely remove any residual wash buffer
5. DNA Elution
Transfer the GeneJET purification column to a clean 1,5 mL tube.
Add 50 µL of Elution Buffer to the center of the GeneJET purification column membrane and centrifuge for 1 minute.
Discard the GeneJET purification column, test with the Nanodrop and store the purified DNA at -20°C.
BioBrick Assembly
Goal
Once you have your constructions on your plasmids, you need to biobrick them before sending them to the registry.
Preparation
Restriction
For restriction you need: distilled water, Bovine Serum Albumin (BSA, used to stabilize restriction enzymes), Restriction Buffer 1,2,3 or 4 (buffer 2 works for all combinations of EcoRI-HF, XbaI, PstI, SpeI), two restriction enzymes per sample.
Determine which restrictions enzymes and Buffer you need using openwetware.org1.
Determine the DNA-concentration of the vector and the insert.
Prepare restriction mix for all restrictions,for each reaction add:
- 12,5 μl od distilled water
- 5,0 μl of Buffer 1,2,3 or 4
- 0,5 μl of BSA
- 1,0 μl of restriction restriction enzyme 1
- 1,0 μl of restriction restriction enzyme 2
Then, for each reaction, add:
- 20 μl restriction mix previously prepared to a PCR-tube
- 1 μg of DNA.
- Distilled water, until your reach a total volume of 50 μl
PCR Purification
Purify the restriction digest, and then determine the concentration of the purified DNA with Nanodrop.
For more information, see the PCR purification protocol.
Ligation
Prepare ligation mix.
Prepare two extra reactions, “No insert” and “No ligase” that will be used as negative controls.
| Per vector/insert | No insert | No ligase | |
|---|---|---|---|
| Ligase | 0,5 μL | 0,5 μL | 0 |
| Vector | |||
| Insert | 0 μL | ||
| Distilled water | |||
| 10X Buffer | 2 μL | 2 μL | 2 μL |
For each reaction,add:
- 50 ng of vector-DNA.
- Insert in three times molar excess, then add 3·50·(length of insert/length of vector) ng of insert-DNA.
- Distilled water, until your reach a total volume of 20 μl
Transformation Test
Transform the ligation product with competent cells, and also "No insert" and "No ligase" controles.- If no colonies form in both controle, the restriction was perfect for both the restriction enzymes.
- If colonies form in both cases, neither of the restriction enzymes worked well. Most of the transformants will contain the vector only.
- If the “No insert” control resulted in many colonies but “No ligase” did not, one of the enzymes worked well and the other did not. Most of colonies will carry the self-ligated vector.
Re-isolate colonies.
Check that colonies have been successfully transformed (e.g. by PCR).
Inoculate those colonies in overnight culture for further use.
Send your part to iGEM registry !
Reference:
- http://openwetware.org/wiki/Enzyme_selection_for_BioBricks_digest
BioBricks Characterization overview

TECAN Analysis
Goal
The Tecan is a plate reader that is able to characterize our bacteria of interest by measuring, for example, growth rate (OD) and/or protein production (GFP). The software saves the measurements over time, thus allowing to study the kinetics of the studied phenomena. With a 96 wells plate (8x12), we are able to test various conditions.
Steps
The hardest part when you use a plate reader to characterize your parts is to carefully choose your controls and anticipate the timings of your bacterial pre-cultures. We tested various controls during this summer:
- TOP10 growth control
- BL21 growth controlFor GFP expression: GFP expression under the control of a pLac promoteLac operator in a delta-LacI strain and GFP expression under the control of a Fur operator in a Δ-Fur strain.
Preparation
Medium preparation
LB medium emit a side signal. As turbidity (OD) and fluorescence of our sample are measured, M9 medium is use instead.
Composition for 50 mL of:
| Reagent | M9 medium (without iron) | M9 medium (with iron) |
|---|---|---|
| M9 salt (5X) | 10 mL | |
| CaCl2 (1M) | 5 µL | |
| MgSO4 (1M) | 100 µL | |
| Glycerol (50%) | 800 µL | |
| Thiamine | 5 µL | |
| NaOH (pH 7.4) | 12.5 µL | |
| H2O | 40 mL | 39 mL |
| FeSO4 (10mM) | - | 50 µL |
| Casamino acids (0.2%) | - | 1 mL |
Once the mixture is prepared, the medium must be filtered to be sterilised using 0.22 µm filter.
Pre-culture preparation
In a 15 mL tube, add 2 mL of M9 medium and inoculate BL21 cells (expression strain) from glycerol
To inhibit the expression of sfGPF, use M9 with iron, instead of classical M9.
After one night of culture, refresh the precultures by diluting them 200 times in M9 medium (with iron and carbenicillin).
After 8 hours of culture, prepare your wells plate according to the following scheme:

Technical triplicate are made to see the variation induced by the manipulator.
Biological duplicate are made to determine the natural variation of the process observed.
Microfluidic experiment
Goal
The aim of microfluidic analysis is to trap bacteria in a chip, with a continuous medium. Hence, the behaviour (e.g. GFP production) of bacteria in exponnential phase can be analyse with microscope during long acquisition (>24h).
Chip preparation
(designed by Boris Kirov)
Mix gently 36 g of Polydiméthylsiloxane (PDMS,condensed formula: (C
Put in a 50 mL tube and centifugate it at 13 000 for 1 minute to remove bubbles from the mixture.
Pour the mixture on the chip mould and put it under a vacuum pump.
Once there is no more bubbles that go out of the mixture, put it in the oven at 80°C for 2 hours.
Separate the different chips and make the holes of the input and outpout of channels with a punch.
Remove all the dust on chips surfaces. Then put chips and glass slides on plasma oven.
After that, put quickly the chip on glass slide and put them in the oven at 80°C for 2 hours.
Bacteria preparation
Make 30 mL of preculture of your strain of interest in LB medium with the recquired antibiotics to avoid contamination.
Let it growth overnight.
Centrifugate and resuspend the pellet in 3 mL of LB medium with the recquired antibiotics and mild detergent
Inject bacteria in the chip with needle and syringe.

Prepare 2x25 mL of LB medium with antibiotic and mild detergent and inject it in the chip.
Check your preparation with the microscope and let the bacteria growth overnight.

At the top and bottom of the picture, the flow of medium go throught the 2 channels.
Acquisition
Wash the chip with your recquired medium (in our case: M9 with or without iron).
Homologous recombination
Goal
Preparation
Be careful, all recombination protocols should be done at 30°C unless indicated otherwise !Strain Preparation
Transform the PTKred Plasmid1 with the strain of interest.
Start an over night culture with the strain transformed with PTKred at 30°C.
Dilute the overnight culture in a ratio of 1 mL/100 mL and add IPTG (isopropyl β-D-1-thiogalactopyranoside; final concentration of 2 mM) and Spectinomycin (60 mg/mL).
Grow up to OD600: 0.5 then make electrocompetent cells .
Integration
Prepare the PCR fragment to be integrated flanked by 50 bp genomic sequences.
Gel-purify the product and transform with the electrocompetent PTKred strain.
Recover in LB for 2 hours with IPTG.
After 2 hours incubation add the selective drug which should be inserted in the genom (for example kanamycin if you have kan R cassette inserted) and incubate overnight in liquid culture at 30°C.
Next day, culture should be very turbid or the recombination did not work. Plate streak 50 μL of the overnight culture on a plate containing selection drug which in this case is kanamycin and grow overnight at 30°C.
Next day pick a colony and make glycerol stocks
Elimination of PTKred plasmid
Inoculate the strain that you have made by Homologous Recombination in LB overnight with spectinomycin at 30°C.
Adding spectinomycin in this step ensures that the plasmid will not integrate in the genome in a rare occurrence.
Dilute 1/100 and grow at 42°C for 4 hours and then plate sreak 50 μL of the culture on LB plate at 42°C.
Next day, pick colonies and grid plate on LB then spectinomycin and grow at 37°C.
Flip Recombinase and Anti-biotic Resistance Gene
Transform a cured strain with PCP20 plasmid
Grow overnight in LB with Cam at 30°C overnight.
Dilute 1/100 and grow for 3 hours at 37°C.
Streak plate on LB plates and grow over night at 42°C.
Repeat the grid.
Reference:
- http://www.addgene.org/41062/
Siderophore detection
Goal
Once our bacteria is transformed with the plasmid with the Fur Binding Site and Lac I and the two plasmids with Enterobactins genes, we need to check if our system really work. The production of siderophores can be detected visualy with Blue Agar Chrome Azurol S (CAS). Without siderophore in the medium, CAS and Hexadecyltrimethylammonium bromide (HDTMA) complexes with ferric iron, producing a blue color. When a bacteria strain produce siderophore, the medium color change from blue to orange.
Preparation
Protocol adapted from Louden, B.C., Haarmann, D., and Lynne, A. (2011). Use of Blue Agar CAS Assay for Siderophore Detection. 1Blue Dye
Solution 1Dissolve 0,06 g of CAS in 50 mL of distilled water.
Solution 2
Dissolve 0,0027 g of FeCl3-6H20 in 10 mL of 10 mM HCl.
Solution 3
Dissolve 0,073 g of HDTMA in 40 mL of distilled water.
Mix
Mix solution 1 with 9 mL of solution 2. Then mix with solution 3.
Solution should have a blue color.
Autoclave and store in a bottle.
Mixture solution
Minimal Media 9 (MM9) Salt Solution Stock
Dissolve 15 g of KH2PO4, 25 g of NaCl and 50 g of NH4Cl in 500 mL of distilled water.
NaOH Stock
Dissolve 25 g of NaOH in 150 mL of distilled water.
pH should be around 12.
20% Glucose Stock
Dissolve 20 g of glucose in 100 mL of distilled water.
Casamino Acid Solution
Dissolve 3 g of Casamino acid in 27 mL of distilled water.
Extract with 3% 8-hydroxyquinoline in chloroform to remove iron.
Filter with a 0,22 µm millipore.
CAS agar preparation
Add 100 mL of MM9 salt solution to 750 mL of distilled water.
Bring pH up to 6 and dissolve 32,24 g of piperazine-N,N'-bis(2-ethanesulfonic acid) (PIPES); PIPES will not dissolve between pH of 5.
Add 15 g of Bacto Agar.
Autoclave and the cool to 50°C.
Add 30 mL of sterile Casamino acid solution and 10 mL of sterile 20% glucose solution to MM9/PIPES mixture.
Slowly add 100 mL of Blue Dye solution along the glass wall while mixing thoroughly.
Bacteria
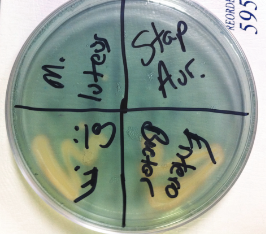
References:
- Louden, B.C., Haarmann, D., and Lynne, A. (2011). Use of Blue Agar CAS Assay for Siderophore Detection. Journal of Microbiology & Biology Education 12,.


