 "
"
Team:Heidelberg/NRPS
From 2013.igem.org



Project

Notebook



Human Practice




Non-Ribosomal Peptide Synthesis. Get to Know the Theory.
Abstract
For many years biological scientists knew of only one direction of genomic-information transfer. Accordingly DNA is transcribed into RNA and RNA is translated into proteins. However, for many organisms this simple assumption turned out to be insufficient to explain their ability to post-translationally modify these complex peptide chains. One of the most striking additional possibilities for bacteria, fungi and even plants to synthesize small peptides is the non-ribosomal peptide synthesis pathway (NRPS).
NRPS are modular mega-enzymes assembled from subunits that are affine for a specific peptide monomer. These enzymes catalyze not only the binding reaction between two monomers but can also add secondary modifications to the associated substrate. Until to now more than 500 different proteinogenic and non-proteinogenic monomers have been found and documented. The consequential assembled small peptide chains often harbor remarkable properties ranging from simple visibility to metal chelating or antibiotic abilities. Even more impressive than the already known wide range of NRPs efficacy is the vast potential of the undiscovered properties that may arise when permuting all these monomers.
Inspired by this idea our team wants to lay the foundation to make this potential accessible to the iGEM community. But first let us have a closer look on the nature of NRPS and learn the basic principles of NRP synthesis.
Even though the ribosome is the molecular machinery mostly used by cells to produce complex polypeptides, bacteria [1, 2] and fungi [3, 4] have an additional pathway that synthesizes peptides via non-ribosomal peptide synthetases (NRPSs). The short peptides assembled by NRPS, termed non-ribosomal peptides (NRPs), comprise a wide range of secondary metabolites, including commonly used antibiotics as well as metabolic and detoxifying enzymes [5, 6]. NRPs range in size from 2-48 amino acid residues [6, 7, 8] and are of remarkable structural variability. In contrast to the ribosomal synthesis pathway which is mainly restricted to the 21 proteinogenic amino acids [9, 10], NRPSs accept more than 500 different monomers as substrates, including non-proteinogenic, N-methylated and D-amino acids [11]. Although the functionality and structure of their synthesized products are of high diversity, NRPS are characterized by a common structural theme: their composition of distinct modular sections (reviewed in [12, 13]). Therefore, the study of NRPSs allows for the exploration of one of the fundamental principles of synthetic biology: modularity.
NRPS Are of Modular Structure
NRPS Are Organized in Biosynthetic Gene Clusters
The genes encoding for NRPS are organized in a large gene cluster [14], so called assembly lines. These contain i) the genes coding for the non-ribosomal peptide synthetases, which can reach a size of up to 1.5 MDa e. g. cyclosporine [15], ii) genes necessary for the biosynthesis of the monomers to be incorporated into the NRP, and iii) tailoring enzymes introducing further modifications in the peptide [16]. Often, the gene clusters code for more than just one peptide synthetase [17]. These co-encoded enzymes are commonly connected by communication domains in order to keep the structure of the assembly line [17]. Thus, the cloning of complete NRPS pathways is facilitated by the clustered structure of the NRPS encoding genes.
One Module - One Amino Acid
Each NRPS is organized in so called modules, where a single module serves as a catalytic unit responsible for the incorporating of one amino acid in the growing peptide chain. The order of the catalytic units defines the sequence and the chemical characteristics of the resulting NRP where the number and order of the units corresponds precisely to the number and order of the amino acids in the resulting peptide [15]. The modules itself are of comprised of smaller subunits termed domains. Each domain fulfills a distinct function in the assembly of the growing peptide chain [13]. The two levels of modularity - modules and domains are shown in Fig. 1.

Figure 1: Modular organisation of NRPS.Non-ribosomal peptide synthetases can be subdivided in modules each incorporating one amino acid. Those are comprised of several different domains, here condensation (C), adenylation (A) and thiolation (T) (according to [13]).
Principle of Non-Ribosomal Peptide Synthesis
For the synthesis of NRPs at least three different domains are essential to carry out the selection of the substrate, its activation and the peptide bond formation: Adenylation, thiolation and condensation domains [13].
Activation of the Thiolation Domains by Phosphopantetheinyl-Tranferases
NRP synthetases contain thiolation domains (T domains) in every module, where the corresponding amino acid is covalently bound to the enzyme via a thioester bond. The essential amino acid residue of the T domains is a serine which is posttranslationally modified to carry the sulfhydryl group required for the thioester bond formation. This modification is carried out by separate proteins - the phosphopantetheinyltranferases (PPTases), which use coenzyme A as cofactor [18]. The reaction is shown in Fig. 2a.
Activation of Monomers by Adenylation Domains
The attachment of the amino acid to the already activated T domain is carried out by the adenylation domain (A domain) which is highly substrate specific for a single monomer. In a first step, the monomer to be incorporated is activated with ATP. Secondly, the thioester bond between the phosphopantetheinyl residue and the monomer's carboxylic acid residue is formed [19]. The reaction is depicted in Fig. 2b.
Formation of Peptide Bonds by Condensation Domains
When two neighboring monomers are activated, the condensation domain (C) catalyzes formation of the peptide bond, which is shown in Fig. 2c. The C domain is selective for the acceptor amino acid and thus the C and A domain of a given module always display the same substrate specificity [20].
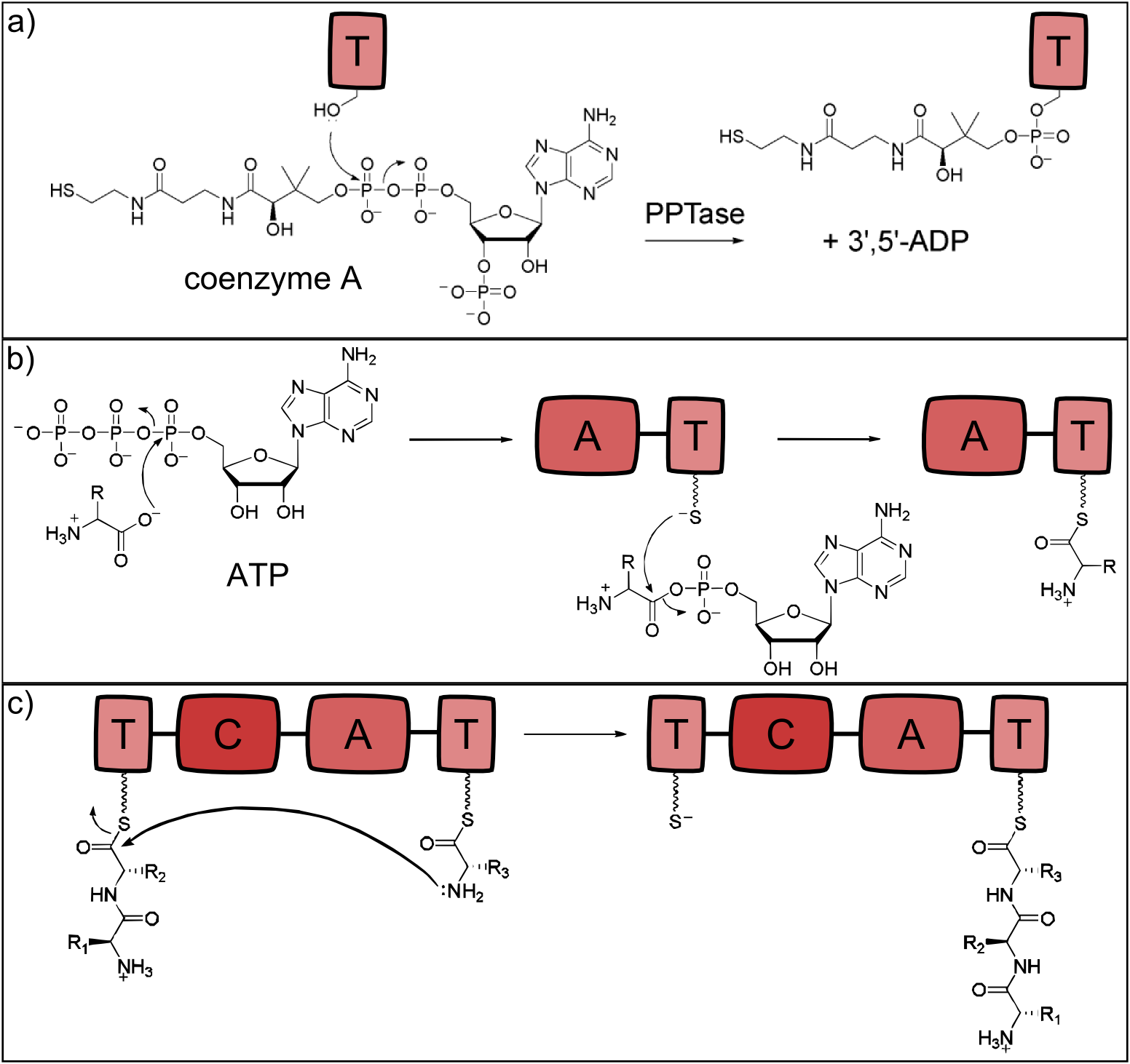
Figure 2: Reactions catalysed by the three basic NRPS domains. a) Thiolation (T) domains are the carrier domains of the monomers and the growing peptide chain. The transfer of phosphopantetheine from coenzyme A to a conserved serine in the T-domain is essential for non-ribosomal peptide synthesis as the thiole residue is necessary for monomer binding. b) The adenylation (A) domain selects the amino acids and covalently binds it to the T domain. It catalyzes two reactions: First, the activation of the monomer by ATP-binding and second, the acylation to the downstream T domain. c) Chain elongation is catalyzed by condensation (C) domains. The C domain enables peptide bond formation between the monomer (acceptor) and the growing peptide chain (donor), resulting in a translocation of the peptide chain to the acceptor T domain (all adapted from [13]).
Termination of NRP Assembly by Thioesterases
In order to terminate the NRP assembly, a thioesterase (TE) domain is located at the end of most NRPSs. TE domains act in two steps. They first bind the peptide to their own conserved serine residue and in a second step this bond is attacked by a nucleophile [21]. The functionalities of the TE domains can roughly be classified into i) product cleavage [22] and ii) macro cyclization [23]. Those TE domains belonging to the former group cleave the thioester bond between the product and the enzyme complex by allowing for a nucleophilic attack with water. TE domains of the second group introduce a macro cycle into the product by an intramolecular nucleophilic attack [21]. This macro cycle can either connect the two peptide termini (yielding a closed ring structure such as tyrocidine [23]) or introduce any other peptide bond based cycle (yielding partially circularized NRPs for instance bacitracin [24]). In addition to those integral TE domains, independent but cluster-encoded thioesterases are known to function as a rescue protein for stalled NRPS [25].
Monomer Modifications
NRPS can incorporate more than 500 different monomers. These monomers include proteinogenic and non-proteinogenic amino acids as well as arylic acids. The latter lack the amine residue and are thus only suitable for chain initiations [11].
Incorporation of D-Amino Acids Is Facilitated by Epimerisation Domains
The most basic derivation is the introduction of D-amino acids into NRPs. Their incorporation is achieved by epimerisation (E) domains located between the T domain of the donor and the C domain of the acceptor module (see Fig. 3). It racemises the donor amino acid and the correct stereo-conformation is selected by the acceptor C domain. Thus the C domains can be classified as either CD or CL specific [26].

Figure 5: A combination of epimerisation (E) and C domains allow for the incorporation of L-amino acids into NRPs. First, the E domain racemises the donor amino acid. Subsequently, a C domain specific for D-amino acids catalyzes the condesation reaction (lower reaction). For the L-stereoconformation, no peptide bond can be formed (upper reaction;adapted from [1]).
Methylation of Monomers by N-Methyltransferases
Another basic modification of monomers is N-methylation carried out by N-methyltransferases (NM). They are located between the A and the T domain of a module and transfer CH3 from S-adenosylmethionine to the amino group of the module's monomer after it has been activated by the A domain [27].
Heterocyclisation of NRPs by Cyclisation Domains
Besides the macro-cyclisation performed by TEdomains, cyclisation (Cy) domains can introduce additional amide bonds between neighboring amino acids within the assembly line. In order to form a heterocycle, serine, threonine or cystein are required as acceptor amino acid [28].
As the borders of the C or A domains can be hard to define, Cy domains and E domains are often displayed as combined C/Cy or C/E-domains. Besides these most common modifications many others can be found within the NRPS cluster or as tailoring enzymes, for instance O-methylations, halogenations, glycosylations or oxidations [13] - there is a lot more to explore!
1. Zhu W, Arceneaux JE, Beggs ML, Byers BR, Eisenach KD, et al. (1998) Exochelin genes in Mycobacterium smegmatis: identification of an ABC transporter and two non-ribosomal peptide synthetase genes. Mol Microbiol 29: 629–639.
2. Byford MF, Baldwin JE, Shiau CY, Schofield CJ (1997) The Mechanism of ACV Synthetase. Chem Rev 97: 2631–2650.
3. Correia T, Grammel N, Ortel I, Keller U, Tudzynski P (2003) Molecular cloning and analysis of the ergopeptine assembly system in the ergot fungus Claviceps purpurea. Chem Biol 10: 1281–1292.
4. Bushley KE, Raja R, Jaiswal P, Cumbie JS, Nonogaki M, et al. (2013) The genome of tolypocladium inflatum: evolution, organization, and expression of the cyclosporin biosynthetic gene cluster. PLoS Genet 9: e1003496.
5. Vining LC (1990) Functions of secondary metabolites. Annu Rev Microbiol 44: 395–427.
6. Zuber P, Nakano MM and Marahiel MA (1993) Peptide antibiotics of Bacilli and other Gram-positive Bacteria. In: Bacillus subtilis and other Gram-positive Bacteria: Physiology, Biochemistry and Molecular Biology. A.L. Sonenshein, R. Losick, and J.A. Hoch (eds.) American Society for Microbiology, Washington D.C., 897-916.
7. Kleinkauf H, Von Dohren H (1996) A nonribosomal system of peptide biosynthesis. Eur J Biochem 236: 335–351.
8. Demain, A. L. In Secondary metabolites: their function and evolution; Symposium, C. F., Ed.. John Wiley & Sons: New York, 1992. Vol. 171.
9. Schwarzer D, Finking R, Marahiel MA (2003) Nonribosomal peptides: from genes to products. Nat Prod Rep 20: 275–287.
10. Lu Y, Freeland S (2006) On the evolution of the standard amino-acid alphabet. Genome Biol 7: 102.
11. Caboche S, Leclere V, Pupin M, Kucherov G, Jacques P (2010) Diversity of monomers in nonribosomal peptides: towards the prediction of origin and biological activity. J Bacteriol 192: 5143–5150.
12. Marahiel MA, Stachelhaus T, Mootz HD (1997) Modular Peptide Synthetases Involved in Nonribosomal Peptide Synthesis. Chem Rev 97: 2651–2674.
13. Fischbach MA, Walsh CT (2006) Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev 106: 3468–3496.
14. Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GS, et al. (2005) Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol 23: 873–878.
15. Marahiel MA (1997) Protein templates for the biosynthesis of peptide antibiotics. Chem Biol 4: 561–567.
16. van Wageningen AM, Kirkpatrick PN, Williams DH, Harris BR, Kershaw JK, et al. (1998) Sequencing and analysis of genes involved in the biosynthesis of a vancomycin group antibiotic. Chem Biol 5: 155–162.
17. Hahn M, Stachelhaus T (2004) Selective interaction between nonribosomal peptide synthetases is facilitated by short communication-mediating domains. Proc Natl Acad Sci USA 101: 15585–15590.
18. Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, et al. (1996) A new enzyme superfamily - the phosphopantetheinyl transferases. Chem Biol 3: 923–936.
19. Mootz HD, Marahiel MA (1997) The tyrocidine biosynthesis operon of Bacillus brevis: complete nucleotide sequence and biochemical characterization of functional internal adenylation domains. J Bacteriol 179: 6843–6850.
20. Bergendahl V, Linne U, Marahiel MA (2002) Mutational analysis of the C-domain in nonribosomal peptide synthesis. Eur J Biochem 269: 620–629.
21. Finking R, Marahiel MA (2004) Biosynthesis of nonribosomal peptides1. Annu Rev Microbiol 58: 453–488.
22. Miller DA, Luo L, Hillson N, Keating TA, Walsh CT (2002) Yersiniabactin synthetase: a four-protein assembly line producing the nonribosomal peptide/polyketide hybrid siderophore of Yersinia pestis. Chem Biol 9: 333–344.
23. Trauger JW, Kohli RM, Mootz HD, Marahiel MA, Walsh CT (2000) Peptide cyclization catalysed by the thioesterase domain of tyrocidine synthetase. Nature 407: 215–218.
24. Konz D, Klens A, Schorgendorfer K, Marahiel MA (1997) The bacitracin biosynthesis operon of Bacillus licheniformis ATCC 10716: molecular characterization of three multi-modular peptide synthetases. Chem Biol 4: 927–937.
25. Schwarzer D, Mootz HD, Linne U, Marahiel MA (2002) Regeneration of misprimed nonribosomal peptide synthetases by type II thioesterases. Proc Natl Acad Sci USA 99: 14083–14088.
26. Luo L, Kohli RM, Onishi M, Linne U, Marahiel MA, et al. (2002) Timing of epimerization and condensation reactions in nonribosomal peptide assembly lines: kinetic analysis of phenylalanine activating elongation modules of tyrocidine synthetase B. Biochemistry 41: 9184–9196.
27. Patel HM, Walsh CT (2001) In vitro reconstitution of the Pseudomonas aeruginosa nonribosomal peptide synthesis of pyochelin: characterization of backbone tailoring thiazoline reductase and N-methyltransferase activities. Biochemistry 40: 9023–9031.
28. Suo Z, Walsh CT, Miller DA (1999) Tandem heterocyclization activity of the multidomain 230 kDa HMWP2 subunit of Yersinia pestis yersiniabactin synthetase: interaction of the 1-1382 and 1383-2035 fragments. Biochemistry 38: 14023–14035.
