 "
"
Team:Heidelberg/Project/Delftibactin
From 2013.igem.org
m |
Nils.kurzawa (Talk | contribs) m |
||
| Line 13: | Line 13: | ||
#tyrocidineText a{ | #tyrocidineText a{ | ||
color:#800000; | color:#800000; | ||
| + | } | ||
| + | |||
| + | p { | ||
| + | text-align:justify; | ||
} | } | ||
</style> | </style> | ||
Revision as of 10:27, 16 October 2013



Project

Notebook



Human Practice




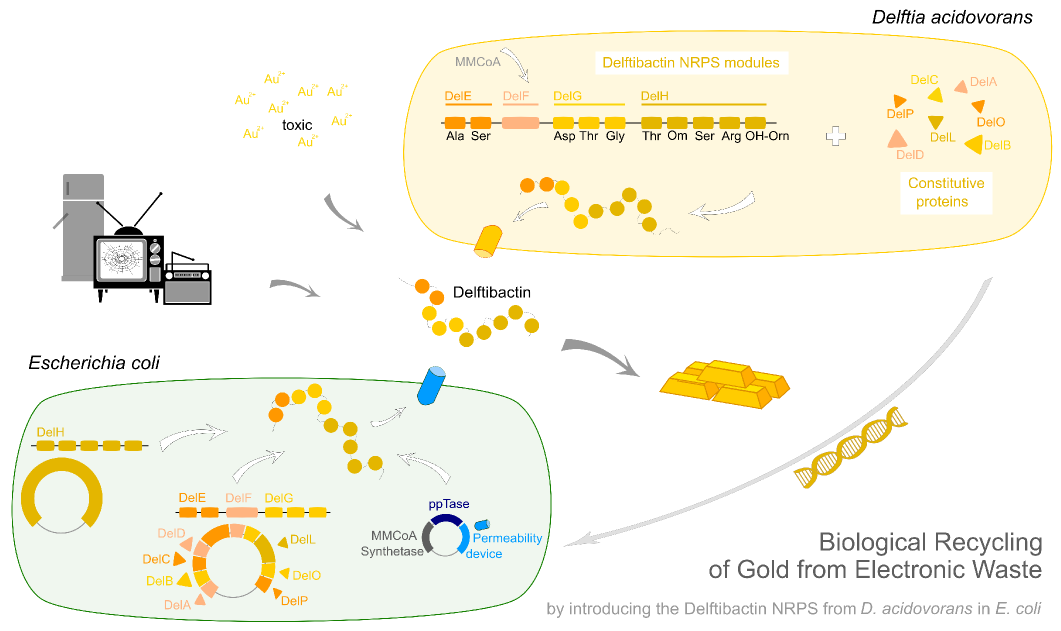
Delftibactin.Recycling Gold from Electronic Waste.
Highlights
- Production and purification of delftibactin, a gold-precipitating NRP, from its native, cultured host Delftia acidovorans.
- Recovery of pure gold from electronic waste by Delftia acidovorans and purified delftibactin.
- Optimization of the Gibson assembly method for the creation of large plasmids (> 30 kbp) with high GC content.
- Amplification and cloning of all components required for recombinant delftibactin production.
- Transfer of the entire pathway from D. acidovorans for the synthesis of delftibactin to E. coli.






Abstract
Efficient recycling of gold from electronic waste using recombinant delftibactin
Undoubtedly, gold is one of the most precious materials on earth. Besides its common use in art and jewelry, gold is also an essential component of our modern computers and cell-phones. Due to the fast turn-over of today’s high-tech equipment, millions of tons of electronic waste accumulate each year containing tons of this valuable metal. The main approach nowadays to recycle gold from electronic waste is by electrolysis. Unfortunately, this is a highly inefficient and expensive procedure, preventing most of the gold from being recovered.
Earlier this year, a publication in Nature Chemical Biology reported the existence of a non-ribosomal peptide – delftibactin - which has the astonishing property to specifically precipitate elemental gold from gold-ion containing solutions. Naturally, delftibactin is produced by Delftia acidovorans, an extremophile bacterium, which secretes delftibactin to complexate and dispose of toxic gold ions present in its environment. Although the exact delftibactin production pathway is not known, bioinformatic predictions claim a non-ribosomal peptide synthesis pathway encoded on a large, 59 kb gene cluster (the del-cluster) to be responsible for delftibactin production.
In this subproject we want to demonstrate that the natural secondary metabolite delftibactin can be efficiently produced in E. coli and used for the recycling of gold from electronic waste. To this end, we developed a cloning strategy based on an optimized Gibson Assembly protocol, enabling the cloning of large, GC-rich genomic regions onto regular low-copy plasmids. We thereby engineered three different plasmids (about 70 kb in total size) enabling the expression of the predicted del-cluster from regular E. coli promoters along with the methylmalonyl-CoA pathway providing the basic delftibactin building blocks and a NRPS activating PPTase, Sfp from Bacillus subtilis.
We want to show that these large constructs can be potentially inserted and expressed by E. coli with the promising perspective that delftibactin could readily be used as an efficient way of gold recycling from electronic waste.
Introduction
The quest for a magical substance to generate gold from inferior metals stirred the imagination of generations. However, this substance, the Philosopher’s Stone, stands for more than just the desire to produce gold. In the old days, the fabled Philosopher’s Stone also represented wisdom, rejuvenation and health. Nowadays, gold is still of great importance for us as it is needed for most of our electronic devices.
In 2007, more than two tons of gold were discarded hidden in electronic waste in Germany. Most of the precious element end up on waste disposal sites as only a minor fraction of 28 % of the gold is recycled also due to the small amounts per devide. Since our planet’s gold supplies are limited, the metal is more and more depleted and the value of gold continously reaches all-time highs. In order to satisfy our society’s need for gold, we have to develop heavy mining techniques involving strong acids, causing devastating impact on human and environment [1] [2] [3].
Besides economical usage of the resource gold, one way to reduce global demands for gold is elevation of gold recovery [4]. Intriguingly, nature itself offers a structure that has been reported to efficiently extract pure gold from solutions containing gold ions. This fascinating molecule is called Delftibactin and is in fact a small peptide secreted by a metal-tolerant bacterium called Delftia acidovorans.
This extremophile has the incredible ability to withstand toxic amounts of gold ions in contaminated soil . What is the special feature of Delftibactin enabling precipitation of gold that efficiently? If one could culture these bacteria and produce Delftibactin in large scales, could one potentially recover gold from electronic waste in a cost- and energy-efficient way? But what is the special feature of Delfibactin to precipitate gold that efficiently?

Delftibactin is no ordinary peptide but a non-ribosomal peptide (NRP) [5] [6]. The efficient and non-polutative large-scale production of this NRP in E. coli could revolutionize the recovery of gold from electronic waste and additionally highlight the plethora of versatile applications for non-ribosomal peptide synthetases (NRPSs). The most sriking feature of these non-ribosomal synthetases is their ablity to incorporate far more than the 21 common amino acids into peptides. They make use of numerous modified and even non-proteinogenic amino acids [7] to assembly peptides of diverse functions.
Delftibactin is a NRP produced by a hybrid NRPS/ polyketide synthase (PKS) system. In their recent publication, Johnston et al. [5] predicted that the enzymes responsible for producing delftibactin are encoded on a single gene cluster, hereafter referred to as Del cluster. It comprises 59 kbp encoding for 21 genes. DelE, DelF, DelG and DelH constitute the hybrid NRPS/ PKS system producing delftibactin, with DelE, DelG and DelH being NRPS and DelF the PKS. The remaining enzymes involved in the Delftibactin synthesis pathway are required for NPRS/ PKS maturation or post-synthesis modification of Delftibactin. The predicted activities of the assumed proteins are:

- DelA: MbtH-like protein, most likely required for efficient delftibactin synthesis [8]
- DelB: thioesterase
- DelC: 4’-phosphopanteinyl transferase: required for maturation of ACP/PCP subunits
- DelD: taurine dioxygenase
- DelL: Ornithine N-monooxygenase
- DelP: N5-hydroxyornithine formyltransferase
We aimed to introduce the large Del cluster into the commonly used, easy-to-culture model organism E. coli to produce Delftibactin. This target bacterium already possesses many components needed for the functionality of non-ribosomal-peptide synthetases. Nevertheless, we introduce nearly the entire Del cluster into E. coli except for DelC (native PPTase). This function is covered by the sfp phosphopanteinyl transferaseintroduced from Bacillus subtilis. As DelF is a PKS, it requires methylmalonyl-CoA as substrate, which is not produced by E. coli. Therefore, the MMCoA synthesis pathway from B. subtilus which is able to activate a wide variety of PKSs including those from Saccharomyces cerevisiae [9] was transferred, too.
The resulting engineered E. coli could be used as host for the delftibactin synthesis pathway, possibly also eliminating the need to introduce DelC. As promoters of the Del cluster were only predicted [10] for Daci_4750 (DelK) and Daci_4760 (DelA) and the cluster is transcribed starting with Daci_4760, we assumed that the entire sequence stretch of approximately 40 kbp is transcribed as single polycistronic mRNA.
Facing these challenges, we decided to approach the project straight forward by cultivation of D. acidovorans and the isolation of native Delftibactin to reproduce the findings of ohnston et al. [5]
Experiments
Our aim is to express delftibactin in E. coli. This will be achieved by introducing three different plasmids which contain parts of the delftibactin-cluster [File:Del cluster.gb] ,a Methylmalonyl-CoA pathway, a Pptase which replaces the DelC-function and a permeability device for the export of the desired NRP.

- Methylmalonyl-CoA, ppTase & permeability device
- DelH
DelA-P - The rest of the genes of the Del-cluster
- Our first aim was to achieve a genomic integration of the genes that encode for components of the Methylmalonyl-CoA pathway into E. coli. The presence of this pathway is required for the production of NRPs. Because the genomic integration turned out to be more challenging then expected a new strategy was developed. Therefore, two plasmids were created (pIK2) containing MethylmalonylCoA amplified from Streptomyces coeliolor and a ppTase amplified from Bacillus subtilis in the Biobrick Backbone pSB3C5 and the permeability device (BBa_I746200) for the outer membrane of E. coli was inserted in another plasmid (pIK1). Team Cambridge revealed in 2007 that Bba_I746200 is toxic. It was itherefore inserted into pIK2 between the two terminators driven by a weak promoter (BBa_J23114) and a weak RBS (Bba_B0030), yielding pIK8 with a total size of 9,467 bp, which was inserted in DH10ß and BL21DE3 via electroporation.
- As the gene encoding DelH alone has a size of 18 kb we decided to clone and introduce this huge gene on a separate plasmid. The first restriction enzyme strategy was problematic because of DelH amplification and the low yield in the ligation. A new GibsonAssembly-strategy was performed and DelH amplified in smaller pieces. It seemed to appear the same problem of as in the pIK1 that E. coli is selecting out the mutated DelH-constructs or is activly mutating it for toxic reasons. A plasmid was designed with the same low copy promotor as in the pIK8 and a low copy RBS [BBa_B0032]. Another shot was a plasmid without promotor so that E. coli has no need to express and mutate DelH. Finally DelH is going to be inserted in E. coli DH10ß and BL21 via electroporation.
DelA-P (the remaining genes of the Del-cluster) [File:Del cluster.gb] was amplified with different primer combinations out of D.acidovorans, and a plasmid was created containing these genes on the pSB4K5 Backbone with lacI promotor and without mRFP. The was transformed into DH10ß and BL21(DE3) via electroporation.
All three plasmid were then electroporated into E. coli BL21 DE3.
Results
Efficient Recycling of Gold from Electronic Waste using Delftibactin
As first, important step in direction of an environmental friendly procedure for recycling gold from gold-containing waste, we wanted to show that the non-ribosomal peptide delftibactin can be used to precipitate gold from gold ion-containing solutions.
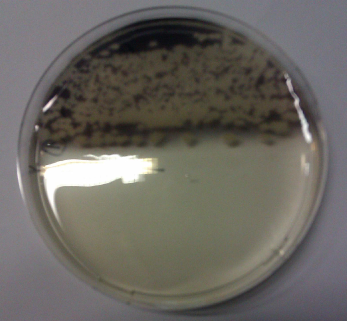
We obtained D. acidovorans DSM-39 from the DSMZ and successfully reproduced the paper by Johnsson et al. [5]. In our experiments, precipitation on agar plates worked even better than described in the paper as shown in Figure 1. D. acidovorans is capable to precipitate solid gold from gold chloride solution as purple-black nanoparticles. Already at low concentrations of gold chloride, gold nonaparticles are precipitated increasing with concentration of gold chloride in solution (Fig. 2).
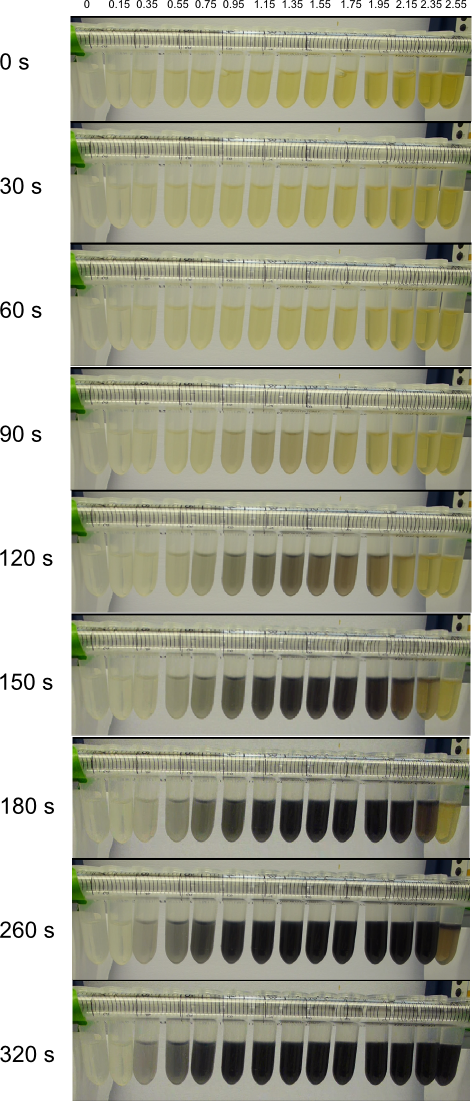
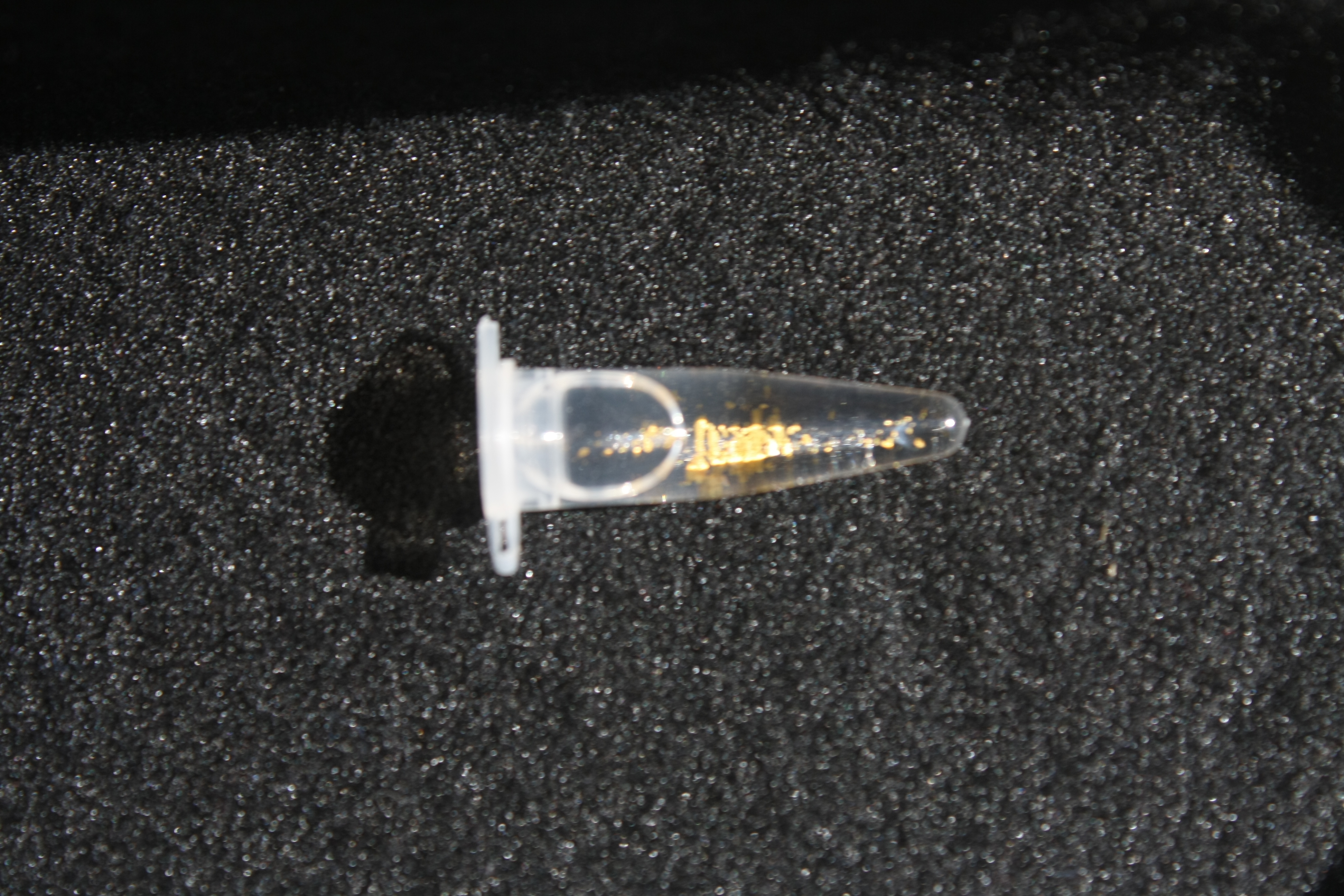
Using supernatants from the new Delftia acidovorans strain SPH-1, we showed precipitation of gold chloride solution to gold nanoparticles. Furthermore, we melted the purple-black nanoparticles to shiny solid gold as shown in figures 6 to 8.


Next, we established purification of Delftibactin using HP20 resins. Additionally, we proved precipitation of gold by the purified Delftibactin (figure 6) and detected it by Micro-TOF.

 Figure 6: Test if purified Delftibactin (diluted 1:10 in H2O) is able to precipitate gold. From left to right: water, 1:10 ACM media, 1:10 supernatant D. acidovorans, 1:10 filtered supernatant D. acidovorans, 1:10 purified Delftibactin |
An important prerequisite for efficient gold recycling from electronic waste is, undoubtedly, that gold precipitation should occur even if the concentration of the gold ions in solution would be rather low, as this would likely be the case for most gold solutions derived from electronic waste.
Therefore, we used an old, broken CPU and established a protocol for dissolving gold from gold-containing metal waste. We incubated golden CPU pins in aqua regia resulting in a gold-ion containing solution (figures 8 to 10).
 Figure 8: Pins removed from an old CPU. |
 Figure 9: Green solution of dissolved pins. |
 Figure 10: Gold chloride solution obtained from an old CPU. |
Moreover, we showed precipitation of dissolved gold recovered from electronic waste by D. acidovorans.
 Figure 11: "Dissolved electronic waste" and D. acidovorans. |
 Figure 12: "Dissolved electronic waste" on D. acidovorans |
 Figure 13: Precipitated "dissolved electronic waste" and D. acidovorans |
Adding this solution to D. acidovorans agar plates resulted in the formation of solid gold nanoparticles.
Taken together, we have successfully established a method enabling the recycling of pure gold from electronic waste using delftibactin produced by D. acidovorans.
Although the recycling was been working efficiently in our hands, the approach of using the natural D. acidovorans bacterial strain for delftibactin production on a larger scale has several disadvantages:
- a) D. acidovorans are relatively slow in growth (colony formation on plates occurs after 2-3 days)
- b) Efficient production of delftibactin requires the strain to be grown in ACM media, which is rather expensive compared to typical E. coli growth media, making the procedure less economic
Therefore, we wanted to engineer an E. coli strain producing delftibactin in high yields, thereby circumventing the abovementioned limitations. To this end, we first had to develop a thorough cloning strategy which would allow us to clone all necessary genes encoding the delftibactin-producing non-ribosomal peptide synthetases and polyketide synthetases from the del cluster (about 50 kb in total) and express them in E. coli alongside with the MethylMalonyl-CoA pathway providing one of the basic substrates in this pathway not naturally present in E. coli.
Amplification of the Del Cluster Genes
The first step towards introducing the Delftibactin expression pathway into E. coli was the amplification of the Del-cluster encoded on the genome of Delftia acidovorans. To this end, we designed Gibson primers and amplified the genes of the non-ribosomal peptide synthetase and the polyketide synthase (PKS) pathway as well as of additional proteins, which were predicted to be necessary for the production of Delftibactin [5]. In the first weeks, PCRs were successfully established and optimized. At the same time, a separate plasmid was created encoding the PPTase from Bacillus subtilis, the MethylMalonyl-CoA pathway and a permeability device [BBa_I746200] [http://parts.igem.org/wiki/index.php?title=Part:BBa_I746200] for the export of the synthesized delftibactin NRP. Additionally, low-copy plasmids from the partsregistry [http://parts.igem.org/Main_Page] were successfully transformed and amplified using corresponding Gibson primers in order to generate the destination backbone fragments for the assembly.
Details of our cloning strategy are shown in figure 14. Notably, the three plasmids we wanted to assemble are huge (23, 32 and 10 kbp in size) and was further complicated due to the high GC content and presence of repetitive elements in Del genes.
Gibson Assembly & Transformation
Assembly of plasmids above 20 kb in size and composed of multiple different fragments is challenging when using conventional restriction-enzyme based cloning. Thus, we have used the Gibson Assembly method [1], which was introduced to the iGEM community by Cambridge in iGEM 2010 as powerful alternative to such common cloning procedures. The assembled constructs of up to 32 kbp in size were transformed into E. coli via electroporation. Correct assemblies of the fragments was tested by analytical restriction digests. The exemplary restriction digest shown above (Fig. 16) confirmed the correct assembly of the three desired constructs as it shows the expected band pattern expected from in silico digestion. Clones (6 and 9) contained the plasmid that encodes for the Methylmalonyl-CoA pathway (Fig. 16a). The obtained DNA sequences were sent in for sequencing over the Gibson-assembled regions for confirmation. [http://www.mwg-biotech.com/],[http://www.gatc-biotech.com/de/index/.html ] The sequencing confirmed the accuracy of the sequence.

The successful generation of DelRest plasmid was proven by different enzymatic restriction digests (Fig. 16b) and also attested by the sequencing. The sequence was compared with the available D. acidovorans SPH1 reference sequence of the Del cluster obtained from NCBI [http://tools.neb.com/] (For further information please visit our labjournal). This shows, that Gibson assembly is a suitable cloning approach for rapid assembly of large NRPS and PKS expression constructs.
Analytical restriction digest of DelH (Fig.16c) also gave rise to a number of positive clones. However, in contrary to the two successfully assembled and sequenced plasmids discussed above, the sequencing results derived from all DelH clones showed various mutations, which were mostly located within the region of the first DelH forward primer. Most of these mutations were deletions present at the very beginning of the DelH coding region. Interestingly, one specific deletion in the DNA sequence was found consistently in several independent clones. As we did not have any clone without mutations, we proceeded with the a DelH clone (termed C5), as this clone did not have any bp deletion but only harbored a minor base-pair substitution (leading to conversion of the DelH Alanine at position 10 to Threonine). Some exemplary sequences are listed below (Tab. 1) of Delftia acidovorans and two observed mutations in different DelH clones.
Tab.1 DelH 5’ sequence, in which most mutations were observed. The ATG start codon is depicted in bold. The table shows the sequence comparison between the DelH reference strand of D. acidovorans and two different exemplary E. coli clones transformed with the plasmid pHM04 (assembled DelH expression vector). The second line shows the accumulated deletion and the third line shows the clone containing 'only' single base pair substitution. Deletions appeared quite frequently while a substitution was only found in a single clone C5. The substitution changes the corresponding Alanine codon to Threonine.
| Organism | Plasmid containing | DNA -Sequence | Conclusion |
|---|---|---|---|
| D. acidovorans | none | ATG GACCGTGGC CGCCTGCGC CAAATCGC | correct |
| E. coli DH10ß | pHM04 | ATG GACCGTG-C CGCCTGCGC CAAATCGC | deletion |
| E. coli DH10ß C5 | pHM04 | ATG GACCGTGGC CGCCTGCGC CAAATCAC | substitution |
Due to the fact that E. coli seemed to somehow selected for mutated DelH clones, we hypothesize that expression of DelH in absence of the other proteins might be toxic for the cells. This would explain why E. coli selects for mutated, none functional, truncated DelH proteins. The same phenomenon of frequent mutations in presumably positive clones was also observed when we started cloning of the permeability device used in the pIK8 construct. The sequenced plasmids showed an unusually high accumulation of mutations compared to other constructs. In case of the methylmalonyl-CoA plasmid (pIK8), the problem was solved by the usage of a weak promoter and a weak ribosome binding site from the partsregistry for driving the expression of the permeability device. Based on this knowledge, DelH is currently being re-assembled into a new backbone (pSB6A1) [http://parts.igem.org/Part:.pSB6A1?title=Part:pSB6A1 ] containing a weak promoter BBa_J23114 [http://parts.igem.org/Part:BBa_J23114 ] and the ribosome binding site BBa_B0032[http://parts.igem.org/Part:BBa_B0032 ]. While the new plasmid is constructed, the following experiments were performed with the C5 clone as we hypothesized, that pHM04 construct #C5 bearing the single nucleotide exchange at position 28 might still show expression of functional DelH when transformed into E. coli (the corresponding amino acid exchange is located at the N-terminus).
Therefore, we transformed all three plasmids, namely the pHM04 #C5 (encoding DelH), the DelRest plasmid (encoding all other del genes despite delH) and the pIK8 construct (bearing the MethylMalonyl-CoA pathway, the Sfp PPTase and the permeability device expression cassette) into E. coli DH10ß and E. coliBL21 DE3 in parallel. This should lead to E. coli cells producing delftibactin and secreting it into the media.
Expression of the Delftibactin NRPSs & Associated Genes
We carried out experiments to test whether our constructs enable expression of the delftibactin NRPS/PKS pathway in E. coli.

For expression of DelH and DelRest, we conducted SDS-PAGE followed by Coomassie stainig. As negative controls we used untransformed cells and cells transformed only with the original plasmid backbones. The proteins DelE, DelG and DelH are significantly larger than any protein that is expressed by our host E. coli. Therefore, the expression of the introduced genes was clearly visible on the SDS-PAGE (Fig. 17). Even though the expression was weak, as we have expected for such a large proteins, clear distinct bands at the expected size of DelE and DelG were detected for the clone transformed with the DelRest plasmid and a band at the size of DelH for the clone transformed with the DelH plasmid. As the promoter in front of DelE and DelG controls the expression of DelA, DelB, DelC, DelD and DelF, too, one can assume simultaneous expression of these Del proteins.

counterclockwise, starting top right: 8 µl, 4 µl, 2 µl, 1 µl bacitracin
For proving that E. coli also produces the permeability device, which is needed for export ofDelftibactin out of the cells, a Hemmhof agar diffusion test with bactracin was performed. Bacitracin is a very large antibiotic which is usually not able to diffuse across the cell membrane passively. Absent growth upon application of bacitracin of bacteria containing the plasmid while in the control cells without the device were not affected by the antibiotic (Fig. 18) confirms expression of the transporter.
In conclusion, we successfully expressed the recombinant Delftibactin NRPS/ PKS pathway as well as the required Methylmalonyl-CoA pathway, the PPTase and permeability device in E. coli.
We furthermore showed, that it is not only possible to assemble large plasmids (in sum these were about 64 kpb in total size in our case) and transform them into E. coli, but showed successful expression of large NRPS/PKS modules in our host strain.
Discussion and outlook
Motivation
Delftibactin is a secondary metabolite naturally produced by Delftia acidovorans and has the ability to specifically precipitate gold from solutions. Today, electronic waste has become an immense environmental problem not only in developed nations, but also third-world countries through the increasing export of waste and disposal challenges.
Therefore, an efficient method to recycle electronic waste is urgently needed. From our work we concluded, that delftibactin could be used as an efficient substance to recycle gold. In fact, many different electronic scraps contain considerable gold amounts such as PC mainboards (566 ppm in terms of weight) and mobile phones (350 ppm). And those electronic devices are more and more demanded by society though their average lifetime seems to decrease steadily. To date, existing methods for gold recycling and safe disposal of electronic waste are still very energy-consuming and harmful to the environment.
Nowadays, harsh substances, such as strong acids, are conventionally used for leaching gold from electronic waste. Applied chemicals pose a potential thread to the environment as they could contaminate the biosphere. Biological agents could become an alternative to chemical clearance of metals from electronic waste. Nature's gold-altering microbes are non-pollutive and they are not even genetically modified. While bacteria such as Cupriavidus metallidurans convert the dissolved element to its metallic form inside the cell [11], species like Chromobacterium violaceum [4] or Delftia acidovorans [5] secrete substances to the surrounding medium for gold precipitation. These microorganisms can therefore be engineered to increase the yield of bioleaching substances.
Achievements
In our experiments, we successfully managed to dissolve electronic waste in aqua regia (i.e. nitro-hydrochloric acid) and neutralize the solution to receive gold chloride. When we added the supernatant of a D. acidovorans liquid culture to the gold chloride solution, pure gold was precipitated. Furthermore, we were able to melt the precipitated gold resulting in little gold flakes. The procedure already worked on a small scale in our project. With increased efforts, yields of Delftibactine for industrial applications are conceivable. In those dimensions, gold could easily be recycled. With Delftibactin there is no further need for chemical reducing reagents to purify gold from solution.
Nevertheless, the efficiency of our approach still has to be improved. We were able to apply Delftibactin for the extraction of gold from electronic waste but still had to expose the CPUs with aqua regis to bring gold in solution. Ideally, one should get rid of the use of this highly corrosive mixture.
To further increase the efficiency of Delftibactin production, we aimed at transferring the entire synthesis machinery for Delftibactin into E. coli. As the NRPS producing delftibactin is a very large enzyme complex consisting of many modules, amplification, cloning and transformation of the constructs was very challenging. Nonetheless, we successfully managed to amplify, assemble and transform all of the genes (in sum 54 kbp) required for production of Delftibactin. In this process, we established protocols, including Gibson assembly of large fragments, and transformation of those constructs into E. coli via electroporation. Optimized procedures will ease the cloning of large customized NRPSs in future.
Challenges
A hurdle to overcome is that the DelH displayed an above-average mutation rate when present in E. coli without the rest of the Delftibactin pathway genes. DelH could potentially be toxic to the E. coli. Therefore, cells select for mutations that render the clones unable to express a funtional DelH protein [12]. In our experiments, the putative selection pressure towards nonfunctional DelH was manifested in deletions, insertions or substitutions in the respective ribsosome binding side or at the beginning of DelH. However, we managed to obtain a single clone, which only possessed a point mutation that potentially does not interfere with protein expression since no false stop codon was created and no frame shift was caused. In this clone, DelH was successfully expressed, yet with an amino acid exchange (Alanine to Threonine) at the N terminus of the protein. At the moment, we are aiming to obtain the gene without any mutation by lowering protein expression. To this end, the most-promising approach is to design a DelH construct containing a weak promoter and weak ribosome binding side. This would decrease the selection pressure caused by DelH. An alternative strategy would be the construction of DelH without any promoter or ribosome binding site. As soon as we would get a mutation-free DelH fragment,,it could be ligated into a plasmid backbone via conventional restriction enzyme-based cloning. A weak promoter and ribosome binding side would keep the detrimental impact of DelH as low as possible. Despite the point mutation, we were able to show that DelH is expressed in E. coli. Our prediction of the tertiary structure of DelH indicated that the.substituted amino acid in the mutant DelH is located at the outer side of the protein. Nevertheless it could still have an important function. Therefore it is not possible to foresee whether the mutation would have any effect on the function of DelH. However alanine is an unpolar amino acid whereas threonine is polar. If the mutation was in a functional region, this could have significant effects rendering DelH nonfunctional but keeping E. coli alive.
1. Eisler R, Wiemeyer SN (2004) Cyanide hazards to plants and animals from gold mining and related water issues. Reviews of environmental contamination and toxicology: 21–54.
2. Eisler R (2004) Arsenic hazards to humans, plants, and animals from gold mining. In:. Reviews of environmental contamination and toxicology. Springer. pp. 133–165.
3. Donato DB, Nichols O, Possingham H, Moore M, Ricci PF, et al. (2007) A critical review of the effects of gold cyanide-bearing tailings solutions on wildlife. Environment international 33: 974–984.
4. Tay SB, Natarajan G, bin Abdul Rahim MN, Tan HT, Chung MCM, et al. (2013) Enhancing gold recovery from electronic waste via lixiviant metabolic engineering in Chromobacterium violaceum. Scientific reports 3.
5. Johnston CW, Wyatt MA, Li X, Ibrahim A, Shuster J, et al. (2013) Gold biomineralization by a metallophore from a gold-associated microbe. Nature chemical biology 9: 241–243.
6. Strieker M, Tanović A, Marahiel MA (2010) Nonribosomal peptide synthetases: structures and dynamics. Current opinion in structural biology 20: 234–240.
7. Caboche S, Leclère V, Pupin M, Kucherov G, Jacques P (2010) Diversity of monomers in nonribosomal peptides: towards the prediction of origin and biological activity. Journal of bacteriology 192: 5143–5150.
8. Baltz RH (2011) Function of MbtH homologs in nonribosomal peptide biosynthesis and applications in secondary metabolite discovery. Journal of industrial microbiology & biotechnology 38: 1747–1760.
9. Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, et al. (1998) Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry 37: 1585–1595.
10. de Jong A, Pietersma H, Cordes M, Kuipers OP, Kok J (2012) PePPER: a webserver for prediction of prokaryote promoter elements and regulons. BMC genomics 13: 299.
11. Reith F, Etschmann B, Grosse C, Moors H, Benotmane MA, et al. (2009) Mechanisms of gold biomineralization in the bacterium Cupriavidus metallidurans. Proceedings of the National Academy of Sciences 106: 17757–17762.
12. Cárcamo J, Ravera MW, Brissette R, Dedova O, Beasley JR, et al. (1998) Unexpected frameshifts from gene to expressed protein in a phage-displayed peptide library. Proceedings of the National Academy of Sciences 95: 11146–11151.
