 "
"
Team:Heidelberg/Project/Indigoidine-Tag
From 2013.igem.org
| Line 159: | Line 159: | ||
==Showing inter-species module compatibility by fusion of Tyrocidine modules to the Indigoidine synthetase== | ==Showing inter-species module compatibility by fusion of Tyrocidine modules to the Indigoidine synthetase== | ||
| + | <p> | ||
Our module-shuffling approach was confirmed by mass spectrometry. Access to such special technical devices and the referring expertise is demanded for detection of small peptides but often limited. That is why we were thinking of a potential alternative to test for the synthesis of NRPs. We wanted to establish an assay accessible to the majority of the community for validation of the presence of custom peptides. Since the synthetase for indigoidine consists of only a single module, it could serve as a paradigm for the fusion to modules of other NRPSs. The pigment Indigoidine could potentially ease detection of peptides when they are fused to the dye that is visible by eye. | Our module-shuffling approach was confirmed by mass spectrometry. Access to such special technical devices and the referring expertise is demanded for detection of small peptides but often limited. That is why we were thinking of a potential alternative to test for the synthesis of NRPs. We wanted to establish an assay accessible to the majority of the community for validation of the presence of custom peptides. Since the synthetase for indigoidine consists of only a single module, it could serve as a paradigm for the fusion to modules of other NRPSs. The pigment Indigoidine could potentially ease detection of peptides when they are fused to the dye that is visible by eye. | ||
<br /> | <br /> | ||
| Line 164: | Line 165: | ||
</p> | </p> | ||
===Fusing single amino acids to Indigoidine=== | ===Fusing single amino acids to Indigoidine=== | ||
| - | + | <p> | |
First, we combined single modules of the Tyrocidine synthetase with the indC synthetase resulting in three distinct NRPSs producing Asparagin, Valine or Phenylalanine respectively, all tagged with Indigoidine (see <a class="fancybox fancyFigure" title="Fig. 9 Composition of three fusion constructs originating from Tyrocidine (tyc) synthetase modules and the Indigoidine synthetase (indC). First row indicates domains with referring modules. Coloured modules in the three rows below were fused together to create plasmids encoding novel NRPSs (rectangle)." href="https://static.igem.org/mediawiki/2013/9/96/Heidelberg_Fusion_ind_tyc_part1.png" rel="gallery1">Fig. 9</a>). | First, we combined single modules of the Tyrocidine synthetase with the indC synthetase resulting in three distinct NRPSs producing Asparagin, Valine or Phenylalanine respectively, all tagged with Indigoidine (see <a class="fancybox fancyFigure" title="Fig. 9 Composition of three fusion constructs originating from Tyrocidine (tyc) synthetase modules and the Indigoidine synthetase (indC). First row indicates domains with referring modules. Coloured modules in the three rows below were fused together to create plasmids encoding novel NRPSs (rectangle)." href="https://static.igem.org/mediawiki/2013/9/96/Heidelberg_Fusion_ind_tyc_part1.png" rel="gallery1">Fig. 9</a>). | ||
<br /> | <br /> | ||
Revision as of 20:11, 25 October 2013



Project

Notebook



Human Practice





Indigoidine-Tag. Inventing the GFP for NRPs.

Highlights
- Creation of an easily detectable NRPS-Tag using the Indigoidine Synthetase
- Proof of principle for inter-species module shuffling
- Protocol for High-Throughput NRPS-assays: RFC 100 and RFC 99
- Empirical determination of optimal domain boarders for T-domain exchange
- Alteration of enzyme activity by exchange of T-domain
- Proof of functionality for synthetic T-domains
Abstract
An integral characteristic of synthetic biology yet often undermined is the ability to learn fundamental knowledge by systematically perturbing a biological system. Non-ribosomal peptide synthetases (NRPS) are predestinated for such a trial and error approach. Their hierarchical organization into modules and domains offer a unique opportunity to spin around their inherent logical assembly and observe if their functionality is preserved. Following this idea, interchangeability of modules within one pathway has already been proven by both, us (LINK) and several research groups (LINK). However, detection of the product, as well as high-throughput screening of functionality remained complicated. Indigoidine, a blue pigment which is synthesized by the NRPS IndC from Photorhabdus luminescens laumondii TT01 (DSM15139), was established as a novel and application-oriented tag functionally fused to synthesized peptides. Streamlining this process demonstrates a substantial reinforcement of the NRPS-Designer, our software tool, and offers the possibility to evaluate and optimize synthesis of Non-Ribosomal Peptides (NRPs) in a high-throughput manner (RFC 99). Furthermore, we established an Indigoidine-production assay based on OD measurement of the blue-colored pigment. We thereby could prove the dependence of the efficiency on the T-domain and the 4'-Phosphopanthetheinyl-transferases (PPTases), resulting in different levels of Indigoidine synthesis.








Introduction
Start introduction here ...
Results
Showing inter-species module compatibility by fusion of Tyrocidine modules to the Indigoidine synthetase
Our module-shuffling approach was confirmed by mass spectrometry. Access to such special technical devices and the referring expertise is demanded for detection of small peptides but often limited. That is why we were thinking of a potential alternative to test for the synthesis of NRPs. We wanted to establish an assay accessible to the majority of the community for validation of the presence of custom peptides. Since the synthetase for indigoidine consists of only a single module, it could serve as a paradigm for the fusion to modules of other NRPSs. The pigment Indigoidine could potentially ease detection of peptides when they are fused to the dye that is visible by eye.
Thus we wanted to investigate whether it is possible to fuse Indigoidine as a tag to NRPs of other pathways even originating from other species. In the following, we will showcase the extent of NRPSs’ modular compatibility by creating inter-species module-fusions between the Indigoidine synthetase from P. luminescens and modules of the Tyrocidine synthetase from B. parabrevis. In those fusion NRPs, Indigoidine serves as a tag that eases identification of E. coli clones synthesizing the customized peptide.
Fusing single amino acids to Indigoidine
First, we combined single modules of the Tyrocidine synthetase with the indC synthetase resulting in three distinct NRPSs producing Asparagin, Valine or Phenylalanine respectively, all tagged with Indigoidine (see <a class="fancybox fancyFigure" title="Fig. 9 Composition of three fusion constructs originating from Tyrocidine (tyc) synthetase modules and the Indigoidine synthetase (indC). First row indicates domains with referring modules. Coloured modules in the three rows below were fused together to create plasmids encoding novel NRPSs (rectangle)." href=" " rel="gallery1">Fig. 9</a>).
" rel="gallery1">Fig. 9</a>).
To assure compatibility the constructs were designed in such a way that the C domain of the C2 module was always used, given its specificity for Glutamine required for the Indigoidine production. SDS-PAGE showed the expected bands for the expression of the NRP synthetases in the transformed BAPI.
The E. coli strain BAPI was used for expression of the NRPS fusions because it carries the required
PPTase endogenously. All three of the fusion variants turned the colonies blue even before expression induction
with IPTG. The blue pigment thus served a first indicator that peptide synthesis was successful. To further
verify the existence of the fusion peptide, we ran comparative thin-layer chromatography (TLC). The native,
purified Indigoidine ran further than our purified dipeptides suggesting that the amino acids were indeed fused
to the pigment (
<a style="float:right" class="fancybox fancyFigure" title="Fig. 10 Comparative TLC of our tagged NRP Val-Ind
with native Indigoidine" href=" " rel="gallery1">
Fig. 10
</a>
). The peptides were detected under visible and UV light due to Indigoidine’s properties as a dye.
" rel="gallery1">
Fig. 10
</a>
). The peptides were detected under visible and UV light due to Indigoidine’s properties as a dye.
We sent a sample of our purified Val-Ind NRP and purified Indigoidine to the mass spectrometry
facility at the Institute for Chemistry handling these samples the same way as the samples sent from the Module
Shuffling experiments. (<a href="https://static.igem.org/mediawiki/2013/7/72/Heidelberg_MS_Val_Ind.pdf"
title="wikilink">Val-Ind MS</a> and <a href="MS_Ind_negativecontrol.pdf" title="wikilink">Negative Control
Ind</a>)
<a class="fancybox fancyGraphical"
href="" style="float: left;
width:150px">
<img style="float:right; width:150px; margin-bottom:10px; padding:1%;border-style:solid;border-
width:1px;border-radius: 5px;" src="">
</img>
<figcaption>Fig. 9 Composition of three fusion constructs originating from Tyrocidine (tyc)
synthetase modules and the Indigoidine synthetase (indC).</figcaption>
</a>
<a class="fancybox fancyGraphical" href="http://igem2013.bioquant.uni-heidelberg.de/wiki/images/7/73/TLC_5.png"
style="float: left; width:150px">
<img style="float:right; width:150px; margin-bottom:10px; padding:1%;border-style:solid;border-
width:1px;border-radius: 5px;" src="http://igem2013.bioquant.uni-heidelberg.de/wiki/images/7/73/TLC_5.png">
</img>
<figcaption>Fig. 10 Comparative TLC of our tagged NRP Val-Ind with native Indigoidine</figcaption> </a>
Using Indigoidine as tag for non-ribosomal peptides
To gather additional evidence for our functional Indigoidine tag, we assembled seven variants with up to four
modules in front of the Indigoidine synthetase (
<a style="float:right" class="fancybox fancyFigure" title="Fig. 11 Composition of seven fusion constructs
originating from Tyrocidine (tyc) synthetase modules and the Indigoidine synthetase (indC). First row indicates
domains with referring modules. Coloured regions in the second row were fused together to create plasmids
encoding novel NRPSs (rectangle)"
href=" " rel="gallery1">
Fig. 11
</a>
) following the same approach as described above.
" rel="gallery1">
Fig. 11
</a>
) following the same approach as described above.
Again the constructs pPW06, pPW09, pPW10, pPW11 and pPW12 turned BAPI colonies blue upon transformation. We
tested the fusion of those peptides by comparative TLC with native Indigoidine. Even with increasing peptide
length, synthesis did not seem to be affected and the dye-properties of the Indigoidine were still preserved (
<a style="float:right" class="fancybox fancyFigure" title="Fig. 12 Comparative TLC of our Tagged NRP with native
Indigoidine" href=" " rel="gallery1">
Fig. 12
</a>
).
" rel="gallery1">
Fig. 12
</a>
).
From this, we deduced that Indigoidine possesses characteristics required for a proper peptide tag
that we would like to propose for use to the community (<a
href="http://dspace.mit.edu/bitstream/handle/1721.1/81333/BBFRFC100.pdf?sequence=1" title="wikilink">RFC 100</a>). For this purpose we have created a ccdB-dependent vector to ease the tagging of NRPs, accessible
through the parts registry. Design of such constructs is enabled with our software, the <a
href="http://igem2013.bioquant.uni-heidelberg.de/NRPSDesigner/" title="wikilink">NRPS Designer</a> .
<a class="fancybox fancyGraphical"
href="" style="float: right;
width:150px">
<img style="float:right; width:150px; margin-bottom:10px; padding:1%;border-style:solid;border-
width:1px;border-radius: 5px;"
src=""> </img>
<figcaption>Fig. 11 Composition of seven fusion constructs originating from Tyrocidine (tyc)
synthetase modules and the Indigoidine synthetase (indC). </figcaption>
</a>
<a class="fancybox fancyGraphical" href=""
style="float: right; width:150px">
<img style="float:right; width:150px; margin-bottom:10px; padding:1%;border-style:solid;border-
width:1px;border-radius: 5px;" src=""> </img>
<figcaption>Fig. 12 Comparative TLC of our Tagged NRP with native Indigoidine. </figcaption> </a>
Experimental validation of software predictions
The NRPS Designer predicts module boundaries and linker regions based on <a
href="http://pfam.sanger.ac.uk/">Pfam</a>. We used these predictions for our module shuffling experiments, which
all worked well in our constructs. However we feel that the borders and linker regions predicted are rather vague
and want to evaluate the predictions to contribute more data to the NRPS Designer. Therefore we systematically
varied the module/ boundaries of the A domain and C domain of C2 within the Val-Ind-construct in relation to the
<a href="http://pfam.sanger.ac.uk/">Pfam</a> prediction. Based on this altered domain positioning, eight
constructs were designed combining narrow, broad and original <a href="http://pfam.sanger.ac.uk/">Pfam</a> module
regions (
<a class="fancybox fancyFigure" title="Fig. 13 Overview over our constructs investigating the different domain
borders in relation to Pfam. Dark colors are variations in the start-region of the A domain borders and light
colors are variations in the end-region of the C domain."
href=" " rel="gallery1">
Fig. 13
</a>
).
). Their functionality was always compared to the original construct based on the boundaries obtained from <a
href="http://pfam.sanger.ac.uk/">Pfam</a>. We could induce the production of Val-Ind in two of our constructs:
pLV03 (narrow and pfam border)and pLV08(broad and narrow border)
" rel="gallery1">
Fig. 13
</a>
).
). Their functionality was always compared to the original construct based on the boundaries obtained from <a
href="http://pfam.sanger.ac.uk/">Pfam</a>. We could induce the production of Val-Ind in two of our constructs:
pLV03 (narrow and pfam border)and pLV08(broad and narrow border)
<a class="fancybox fancyGraphical"
href="" style="float: right;
width:150px">
<img style="float:right; width:150px; margin-bottom:10px; padding:1%;border-style:solid;border-
width:1px;border-radius: 5px;" src="">
</img>
<figcaption>Fig. 13 Overview over our constructs investigating the different domain borders in
relation to Pfam. </figcaption>
</a>
To summarize, with our inner- and interspecies module shuffling, we successfully validated the concept of
modularity. We have integrated our experimental data and fuelled it into our software to improve its prediction
accuracy. To make NRPSs and their custom design more accessible to the community, we have submitted standardized
versions of the modules we used for our shuffling experiments, to the parts registry (see the <a
href="http://parts.igem.org" title="wikilink">parts registry</a> entries).
Conclusion
We can prove all mentioned claims since we have:
- Exchanged modules of of the Tyrocidine synthetase,
- Constructed a custom dipeptide- and a custom tripeptide-NRPS,
- Detected both the engineered NRPSs and the synthetic peptides via SDS-PAGE and mass spectrometry,
- Demonstrated compatibility of modules from the Tyrocidine synthetase of B. parabrevis with modules from the Indigoidine synthetase of P. luminescens and
- Established Indigoidine as functional tag that allows for cost-efficient detection of newly synthesized NRPs
The achievements were enabled through and fed back to the NRPS Designer. This software tool incorporates a database utilizing our experimental on NRPS modules for guiding through user-oriented design of NRPSs.
Methods
Table 1: Bacterial strains and genes of interest derived thereof. The indigoidine synthetase bpsA was kindly supplied by the Fussenegger lab at ETH Zurich.| Strain | Gene | Function |
|---|---|---|
| Photorhabdus luminescens laumondii TT01 DSM15139 | indC | Indigoidine synthetase |
| Streptomyces lavendulae lavendulae | bpsA | Indigoidine synthetase |
| Photorhabdus luminescens laumondii TT01 DSM15139 | ngrA | PPTase |
| Escherischia coli BAP1 | sfp | PPTase |
| Streptomyces verticillus ATCC15003 | svp | PPTase |
| Escherischia coli MG1655 | entD | PPTase |
| Delftia acidovorans SPH-1 | delC | PPTase |
Cloning Strategy
We assembled the different indC variants on a chloramphenicol resistance backbone (pSB1C3) with an IPTG-inducable lac-promoter, the ribosome binding site BBa_B0034 and the coding sequence of the respective indC variant. The indC plasmids should be co-transformed with a PPTase construct to get a significant and fast indigoidine production. Therefore, we used a second plasmid backbone carrying a kanamycin resistance (pSB3K3). We assembled five pSB3K3 derived plasmids, each carrying an expression cassette with an IPTG induceable lac-promotor, the BBa_B0029 ribosome binding site and the coding sequence of the respective PPTase (sfp, svp, entD, delC and ngrA; see Table 1). We used E. coli TOP10 for co-transformations of the possible combination of the indC variants (2) and all PPTase plasmids (5).Circular Polymerase Extension Cloning
Circular Polymerase Extension Cloning (CPEC) is a sequence-independent cloning method based on homologous recombination of double-strand DNA overlaps of vector and insert(s) (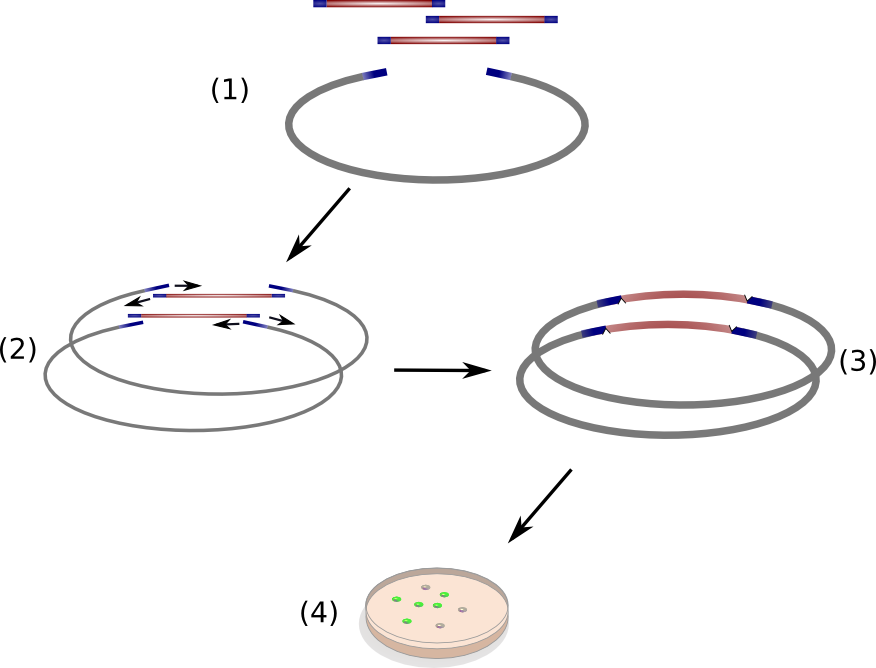
- initial denaturation at 98°C for 30 s
- 5 cycles with:
- denaturation step at 98°C for 5 s.
- annealing step at 53°C for 15 s
- elongation/filling up step at 72°C for 20 s/kbp of longest fragment.
- final extension at 72°C for three times the calculated elongation time.
- (Optional: Hold at 12°C )
After CPEC, 5 µl of of the reaction mixture were used for transformation. The remaining volume was used for quality check on a gel with small pockets (10 to 20 µl in volume). The following primers were used for all CPEC experiments into standard BioBrick backbones: BBa_J04450_stem_loop_fw, Bba_J04450_B0034-RBS_ATG_rv, Bba_J04450_B0029-RBS_ATG_rv. The reverse primers (rv) differ in the ribosomal binding sites they introduce: Bba_J04450_B0034-RBS_ATG_rv contains the ribosomal binding site used in J04450, Bba_J04450_B0029-RBS_ATG_rv introduces the ribosomal binding site B0029 which is of weaker than B0034.
Generation of the ccdB-Ind construct
To minimize the background colonies when exchanging the T-domain of the indigoidine synthetase we generated the ccdB-Ind plasmid where we replaced the indC T-domain with the ccdB gene (Modul structure: AoxA-ccdb-TE) which kills E. coli TOP10 cells but not E. coli OneShot ccdB survival cells. Test-transformation in both E. coli TOP10 and the E. coli OneShot ccdB survival cells showed that background colonies could be eliminated by this strategy (Plattenbild top10 vs survival cells). We used the ccdB-Ind for all further CPEC experiments aiming to swap T-domains. Primers for the backbone CPEC fragments were designed to facilitate the amplification of the entire ccdB-Ind plasmid while omitting the ccdB sequence (compare to Figure??). Assembly of the finale indigoidine synthase products with exchanged T-domain was achieved by CPEC as described or above or HiCT (RFC 99).Examination of T-domain borders
We exchanged the T-domain of indC with the T-domain of bpsA and varied the size of the exchanged DNA sequence, thus examining several domain borders (compare to Figure ??). We used the CPEC assembly method and the indC-ccdB plasmid for this approach.Test of various T-domains from different NRPS modules
For the investigation of additional T-domains from less related NRPS modules, we selected the border combination b31??? which was positive in the test with bpsA. We used the T-domains of the following genes: Table 2: Genes of which T-domains have been extracted and introduced to indC| Gene | Organism | Original function |
|---|---|---|
| entF | Escherichia coli K-12 | NRPS module of enterobactin synthesis pathway |
| tycA1 | Brevibacillus parabrevis | 1st module in tyrocidine synthesis cluster |
| tycC6 | Brevibacillus parabrevis | Last module in tyrocidine synthesis cluster |
| delH4 | Delftia acidovorans SPH-1 | 2nd but last module in delftibactin synthesis cluster |
| delH5 | Delftia acidovorans SPH-1 | Last module in delftibaction synthesis cluster |
| plu2642 | P. luminescens DSM15139 | NRPS of unknown function (one module: A-T-TE) |
| plu2670 | P. luminescens DSM15139 | module of NRPS pathway of unknown function |
All T-domains from the respective genomes were amplified using CPEC primers with a uniform 5’-end and a 3’-end specific for the respective gene. For the assembly of the hybrid-indigoidine synthetases by CPEC, the indC-ccdB construct was used.
Creation of synthetic T-domains
All R scripts used in the following sections are based on R version R-3.0.1. Different assumptions about the evolutionary conservation of T-domains were examined: i) conservation of a specific module across different species, ii) conservation of T-domains across different modules for the same species, iii) conservation of T-domains across different species, iv) conservation of similar modules across different species. According to these three assumptions, different libraries of homologous protein sequences were generated using ncbi protein BLAST (blast.ncbi.nlm.nih.gov) with standard parameters:- query sequence: indC; Search set: non-redundant protein sequences without organism restriction
- query sequence: indC T-domain; Search set: non-redundant protein sequences within P. luminescens
- query sequence: indC T-domain; Search set: non-redundant protein sequences without organism restriction
- query sequences: indC, bpsA, entF, delH5 and tycC6; Search set: non-redundant protein sequences without organism restriction;
The 50 closest related protein sequences contained in each the library were subjected to a multiple sequence alignment (MSA) using clustalO (http://www.ebi.ac.uk/Tools/msa/clustalo/). with standard parameters for protein alignments. For library generation iv), each query sequence was BLASTed separately and the 50 best results of each query were combined i.e. a total of 250 sequences for the MSA. After library generation, the following three methods were employed to design different synthetic T-domains.
Consensus method
Based on the .clustal file obtained from the MSA of the homology libraries, a consensus sequence using the UGENE software (http://ugene.unipro.ru/) with a threshold of 50% was created (i.e. if an amino acid appears in 50% or more of all sequences at a specific position it is considered as a consensus amino acid). For the creation of the synthetic T-domains, this consensus sequence was used to fill the gaps where there was no consensus amino acid with the original amino acid from the indC T-domain. By this approach, T-domains were generated which might deviate from the original sequence at positions with at least average conservation but coreespond to the original one if there is less conservation.
Guided random method
In this approach, the multiple sequence alignments (MSA) generated by the consensus method was used. Implemented in R [Referenz], a position-specific profile was generated which has the same length as the MSA and contains the rate at which amino acids occur at any given position of the sequence alignment. The synthetic T-domain is created by position-wise generation of the sequence where the probability of choosing an amino acid at a given position is determined by the rate in the profile.
Randomized generation method
For generation of synthetic sequences by the randomized generation method, every amino acid was assigned a score of 1 or 0, i.e. occuring at least ones or not at all at a given position in the MSA. In the subsequent generation of the synthetic T-domain sequence of the synthetic domain, any amino acid assigned 1 had the same likelihood of being chosen at this position.
Guided random method
In this approach, the multiple sequence alignments (MSA) generated by the consensus method was used. Implemented in R [Referenz], a position-specific profile was generated which has the same length as the MSA and contains the rate at which amino acids occur at any given position of the sequence alignment. The synthetic T-domain is created by position-wise generation of the sequence where the probability of choosing an amino acid at a given position is determined by the rate in the profile.Randomized generation method
For generation of synthetic sequences by the randomized generation method, every amino acid was assigned a score of 1 or 0, i.e. occuring at least ones or not at all at a given position in the MSA. In the subsequent generation of the synthetic T-domain sequence of the synthetic domain, any amino acid assigned 1 had the same likelihood of being chosen at this position.Seven synthetic T-domains were designed based on differnt combinations of the homology libraries and sequence generation methods.
Table 3: Overview of the homology libraries and sequence generation methods employed for the generation of seven synthetic T-domains| Domain ID | Homology library | Sequence generation method |
|---|---|---|
| synT1 | library i | consensus |
| synT2 | library ii | consensus |
| synT3 | library iii | consensus |
| synT4 | library iv | consensus |
| synT5 | library i | guided random |
| synT6 | library iv | guided random |
| synT7 | library i | randomized generation |
Figure ??shows the multiple sequence alignment of the seven synthetic T-domains and the native indC T-domain. After the generation of the T-domain amino acid sequences, the OPTIMIZER web-tool(http://genomes.urv.es/OPTIMIZER/) was used to obtain the corresponding DNA sequence. E. coli K-12 was set as strain for codon optimization and most frequent was chosen as codon option. The generated DNA sequence was cured from internal RFC10 cutting sites and CPEC cloning overhang required for the T-domain swapping into the ccdb construct were introduced. The synthetic T-domains were ordered at IDT (Integrated DNA Technologies, Coralville, Iowa). In order to obtain sufficient amounts of DNA, the synthetic T-domains were amplified via PCR. IndC-hybrid constructs of the native IndC with exchange of the native T-domain by the synthetic variants were assembled using CPEC and the indC-ccdB construct as backbone. The synthetic T-domains were amplified for CPEC using the same primers as for the native indC T-domain.
Table 4: Overview of primers that MAY be utilized for backbone linearization: The reverse primers (rv) differ in the ribosomal binding sites they introduce: BBa_J04450_B0034-RBS_ATG_rv contains the ribosomal binding site B0034, BBa_J04450_B0029-RBS_ATG_rv introduces the ribosomal binding site B0029 which is weaker than B0034. Note that the 5' overhangs (underlined) of the reverse primers (rv) already include the start codon (depicted in bold) of the coding sequence to be introduced as insert into the corresponding backbone. The resulting expression cassette will be driven by the Plac promoter (R0010).| Primer | Primer sequence(5’ --> 3’) | Cutting site |
|---|---|---|
| BBa_J04450_stem_loop_fw | TAATGA GCTAGC TAATAACGCTGATAGTGCTAGTG | NheI |
| BBa_J04450_B0034-RBS_ATG_rv | CAT GGTACC TTTCTCCTCTTT CTCTAGTATGTGTG | KpnI |
| BBa_J04450_B0029-RBS_ATG_rv | CAT GGATCC GGTTTCCTGTGTGAA CTCTAGTATGTGTGAAATTGTTATCC | NheI |
Quantitative indigoidine production assay
1. OD MEASUREMENT by TECAN plate reader
96-well plates are prepared with 100 μl LB-medium/well containing appropriate antibiotics (chloramphenicol and kanamycin for the indigoidine and PPTase contrcuts, respectively) and each well is inoculated with single colonies (in duplicates) from plates positive for the co-tansformation experiments i.e. from plates with blue colonies. Two sets of negative controls are also inoculated on the plate: First, pure medium serving as the baseline for background correction for the OD measurements. Second, transformation controls accounting for potential differences in cell growth due to expression of proteins contained on the plasmids, i.e. the antibitotic resistance gene and IndC. In this set of controls, the plasmid used in co-transformation with the PPTase plasmid contains IndC-constructs carrying a randomly generated sequence instead of the T-domain. A second 96 well plate was prepared with 180 µl LB-medium/well for the measurement itself. The 96-well plate containing the pre-cultures of the co-transformed colonies was inoculated for 24 hours at 37°C. Subsequently, 20 µl of the pre-culture was transferred to the measurement plate. The absorbance of the bacterial cultures was measured at wavelengths ranging from 400 nm to 800 nm in intervals of 10 nm for each well every 30 min for 30 hours at 30°C in a Tecan infinite M200 plate reader. For the measurement plate, Greiner 96-well flat black plates with a clear lid were used.2. Data analysis
Detecting the amount of the NRP expressed by the bacterial host strain is desirable. By tagging the NRP with indigoidine, the amount of the fusion peptide can be determined by quantifying the amount of blue pigment present in the cells. As the amount of blue pigment is proportional to the amount of the NRP of interest, a method for the quantification of the blue pigment will yield information about the expression of the NRP. Quantification of the pure indigoidine pigment can be easily achieved by optical density (OD) measurements at its maximum wavelength of about 590 nm. In cellular culture, indigoidine quantification by OD measurements is impaired. Cellular density of liquid cultures is standardly measured as the optical density (OD) at a wave length of 600 nm, i. e. the absorption peak of indigoidine interferes with the measurement of cell density at the preferred wave length (compare to Figure 3, grey dashed line). Thus, for measurement of NRP expression without time consuming a priori purification of the tagged-protein, a method to separate the cellular and pigment-derived contributions to the OD is required (compare to Figure 3, brown and blue lines, respectively). The method of choice, as described by Myers et al.[2013], requires the OD measurement of cell culture at two distinct wavelengths: the robust wave length ODR and the sensitive wave length ODS. The concentration of indigoidine will have to be deducted from measurements at ODS = 590 nm: <math> OD_{S,+P} </math> [Indigoidine]= 〖〖OD〗_(S,+P)-OD〗_(S,-P) with 〖OD〗_(S,+P) being the overall OD measurement and 〖OD〗_(S,-P) being the scattering contribution of the cellular components at the sensitive OD. The scattering contribution of the cellular compenents at ODS (ODS,-P ) can be calculated from the scattering contribution measured at the robust wave length according to the following formula: [[File: The correction factor δ is be determined by measuring the OD of pure cellular culture without indigoidine at both the wavelength 〖OD〗_(S,-P) and 〖OD〗_R and calculating their ratio. Finally, the indigoidine production can be determined as [[File:Heidelberg_5.png]] For the calculation of the cellular component when measuring indigoidine producing liquid cell cultures, OD measurement at 800 nm as robust wavelength is recommended. By the approach described above, quantitative observation of the indigoidine production in a liquid culture over time as well as the indigoidine production in relation to the cell growth can be conducted. Background correction i. e. the contribution of the culture medium to the OD measurement is achieved by subtracting the mean of pure culture medium replicates from all OD values measured.



1. Fischbach MA, Walsh CT (2006) Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev 106: 3468–3496.
2. Takahashi H, Kumagai T, Kitani K, Mori M, Matoba Y, et al. (2007) Cloning and characterization of a Streptomyces single module type non-ribosomal peptide synthetase catalyzing a blue pigment synthesis. In:. Vol. 282. pp. 9073–9081.
3. Brachmann AO, Kirchner F, Kegler C, Kinski SC, Schmitt I, et al. (2012) Triggering the production of the cryptic blue pigment indigoidine from Photorhabdus luminescens. In:. Vol. 157. pp. 96–99.
4. Owen JG, Robins KJ, Parachin NS, Ackerley DF (2012) A functional screen for recovery of 4’-phosphopantetheinyl transferase and associated natural product biosynthesis genes from metagenome libraries. In:. Vol. 14. pp. 1198–1209.
5. Doekel S, Marahiel MA (2000) Dipeptide formation on engineered hybrid peptide synthetases. In:. Vol. 7. pp. 373–384.
6. Thirlway J, Lewis R, Nunns L, Al Nakeeb M, Styles M, et al. (2012) Introduction of a non-natural amino acid into a nonribosomal peptide antibiotic by modification of adenylation domain specificity. In:. Vol. 51. pp. 7181–7184.
7. Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C (2001) Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. In:. Vol. 291. pp. 1790–1792.
